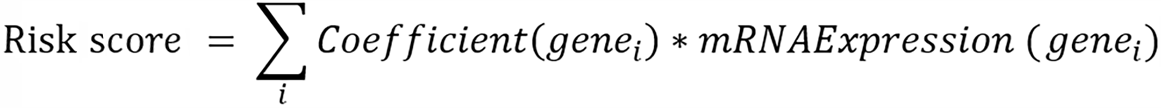Ren QS, Sun Q, Cheng SQ, Du LM, Guo PX. Hepatocellular carcinoma: An analysis of the expression status of stress granules and their prognostic value. World J Gastrointest Oncol 2024; 16(6): 2571-2591 [PMID: 38994142 DOI: 10.4251/wjgo.v16.i6.2571]
Corresponding Author of This Article
Ping-Xuan Guo, Doctor, Department of Anesthesiology, Kailuan General Hospital, North China University of Science and Technology, No. 57 Xinhua East Street, Tangshan 063000, Hebei Province, China. kyguopingxuan@163.com
Research Domain of This Article
Medical Informatics
Article-Type of This Article
research-article
Open-Access Policy of This Article
This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Baishideng Publishing Group Inc, 7041 Koll Center Parkway, Suite 160, Pleasanton, CA 94566, USA
Share the Article
Ren QS, Sun Q, Cheng SQ, Du LM, Guo PX. Hepatocellular carcinoma: An analysis of the expression status of stress granules and their prognostic value. World J Gastrointest Oncol 2024; 16(6): 2571-2591 [PMID: 38994142 DOI: 10.4251/wjgo.v16.i6.2571]
Qing-Shuai Ren, Department of Cardiovascular Surgery, North China University of Science and Technology Affiliated Hospital, Tangshan 063000, Hebei Province, China
Qiu Sun, Department of Hepatobiliary, Kailuan General Hospital, Tangshan 063000, Hebei Province, China
Shu-Qin Cheng, Department of Gastroenterology and Hepatology, Tianjin Medical University General Hospital, Tianjin Institute of Digestive Diseases, Tianjin Key Laboratory of Digestive Diseases, Tianjin 300000, China
Li-Ming Du, Department of Traditional Chinese Medicine, Kailuan General Hospital, Tangshan 063000, Hebei Province, China
Ping-Xuan Guo, Department of Anesthesiology, Kailuan General Hospital, North China University of Science and Technology, Tangshan 063000, Hebei Province, China
Author contributions: Ren QS and Sun Q contributed equally to this work. Ren QS and Sun Q carried out the studies and drafted the manuscript; Ren QS, Sun Q, Cheng SQ, and Du LM analyzed data; Sun Q and Guo PX read and revised the manuscript; and all authors read and approved the final manuscript.
Supported byHebei Traditional Chinese Medicine Scientific Research Project, No. 2023223.
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Data sharing statement: The original contributions presented in the study are included in the article/supplementary files. Further inquiries can be directed to the corresponding authors.
Corresponding author: Ping-Xuan Guo, Doctor, Department of Anesthesiology, Kailuan General Hospital, North China University of Science and Technology, No. 57 Xinhua East Street, Tangshan 063000, Hebei Province, China. kyguopingxuan@163.com
Received: December 19, 2023 Revised: January 24, 2024 Accepted: April 1, 2024 Published online: June 15, 2024 Processing time: 178 Days and 11.7 Hours
Abstract
BACKGROUND
Hepatocellular carcinoma (HCC) is a global popular malignant tumor, which is difficult to cure, and the current treatment is limited.
AIM
To analyze the impacts of stress granule (SG) genes on overall survival (OS), survival time, and prognosis in HCC.
METHODS
The combined The Cancer Genome Atlas-Liver Hepatocellular Carcinoma (TCGA-LIHC), GSE25097, and GSE36376 datasets were utilized to obtain genetic and clinical information. Optimal hub gene numbers and corresponding coefficients were determined using the Least absolute shrinkage and selection operator model approach, and genes for constructing risk scores and corresponding correlation coefficients were calculated according to multivariate Cox regression, respectively. The prognostic model’s receiver operating characteristic (ROC) curve was produced and plotted utilizing the time ROC software package. Nomogram models were constructed to predict the outcomes at 1, 3, and 5-year OS prognostications with good prediction accuracy.
RESULTS
We identified seven SG genes (DDX1, DKC1, BICC1, HNRNPUL1, CNOT6, DYRK3, CCDC124) having a prognostic significance and developed a risk score model. The findings of Kaplan-Meier analysis indicated that the group with a high risk exhibited significantly reduced OS in comparison with those of the low-risk group (P < 0.001). The nomogram model’s findings indicate a significant enhancement in the accuracy of OS prediction for individuals with HCC in the TCGA-HCC cohort. Gene Ontology and Gene Set Enrichment Analysis suggested that these SGs might be involved in the cell cycle, RNA editing, and other biological processes.
CONCLUSION
Based on the impact of SG genes on HCC prognosis, in the future, it will be used as a biomarker as well as a unique therapeutic target for the identification and treatment of HCC.
Core Tip: Hepatocellular carcinoma (HCC) is challenging to cure because the available treatments are ineffective, and this cancer demonstrates a significant degree of resistance to standard chemotherapy and radiotherapy modalities. In cancer cells, the formation of stress granules (SGs) is thought to protect them from apoptosis and induce resistance to anti-cancer drugs or radiotherapy, making SGs a potential target for cancer therapy. Throughout this investigation, we found that signatures of seven SG genes can serve as independent predictors of overall survival in patients with HCC, and survival and prognosis analyses have been conducted for each expression of the hub gene.
Citation: Ren QS, Sun Q, Cheng SQ, Du LM, Guo PX. Hepatocellular carcinoma: An analysis of the expression status of stress granules and their prognostic value. World J Gastrointest Oncol 2024; 16(6): 2571-2591
Liver cancer ranks as the fifth highest prevalent malignancy globally and the fourth primary contributor to cancer-associated mortality[1], with a calculated number of new cases estimated to be 906000, corresponding to 830000 deaths worldwide each year[2]. There are two primary forms of significant liver disorders, cancer-hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma, and less prevalent tumors, including hemangiosarcoma and hepatoblastoma, of which HCC constitutes over 80% of the global incidence of primary liver cancer[3]. Asian countries exhibit the highest prevalence of HCC, which ranks as the second highest prominent cause of death related to cancer, and it accounts for 72.5% of global incidence and 73.3% of global mortality in 2020[4]. HCC can occur in a category of chronic liver disorders, for instance, and non-alcoholic fatty liver disease, alcoholic liver disease, chronic hepatitis B and C virus infection, most of which begin with an abnormal buildup of fat through liver cells, a status known as steatosis and hence fatty liver disease. Over a period of time, hepatocyte death and inflammation are caused by endoplasmic reticulum stress, lipotoxicity, and mitochondrial dysfunction (steatohepatitis) and result in a buildup of fibrotic tissue throughout the liver cells. The progression of unresolved chronic inflammation and fibrosis is observed over a certain amount of time, and regenerative nodules comprising hepatocytes with low differentiation, arise within the parenchyma, ultimately resulting in cirrhosis[1,5]. Most HCC (80%-90%) is preceded by cirrhosis[6]. HCC is challenging to cure because the available treatments are ineffective, and this cancer demonstrates a significant degree of resistance to standard chemotherapy and radiotherapy modalities. The most efficacious approach continues to be surgical resection or liver transplantation. However, not all patients meet the criteria to undergo surgical intervention, with recurrence rates approaching 70% five years after resection[1,7]. Therefore, further understanding of the HCC molecular mechanisms is needed to construct new effective prevention and treatment approaches.
The pathogenesis of HCC is complex, involving multiple molecular malfunctions, including cell cycle dysregulation, chromosomal instability, DNA methylation changes, immune regulation, epithelial-mesenchymal transition, HCC stem cells progression, and the dysregulation of microRNA (miRNA)[8], among these, aberrant gene expression post-transcriptional regulation is the most crucial element that drives the progression of liver carcinogenesis. Alterations in the expression or function of miRNAs and RNA binding proteins (RBPs), or both, can lead to the reduction of mRNA levels or inhibit the translation of transcripts that regulate liver metabolism, inflammation, and carcinogenesis[9]. In the case of gene mutations, alterations in post-transcriptional mechanisms that regulate gene expression have the potential to result in the oncogenes’ overexpression or the tumor suppressors’ under-expression, thus, promoting the progression of cancer. Specific regulatory processes occur within small cytoplasmic ribonucleoprotein (RNP) foci, including stress granules (SGs) or p-bodies, and the mRNA’s destiny is identified (i.e., degradation and translation)[10]. SGs are RNP granules located in the cytoplasm that emerge as a result of stress. These particles are thought to be evolutionarily conserved cellular defense mechanisms that can resist a variety of stresses[11]. During times of physiological stress, SG selectively isolates specific transcripts and proteins from the soluble fraction of cytoplasm, which may be possible to be a mechanism that serves as a response to stress, regulating both gene expression and cell signaling[12]. SGs are dynamically regulated according to cell type and stress, they can influence cell development, apoptosis, and senescence, and their regulation is frequently linked to age-related human disorders, including cancer and neurodegenerative diseases[13]. The overexpression of various components of SGs has been detected in different tumor types, including sarcoma, pancreatic cancer, HCC, and glioblastoma[14]. Given that cancer cells are naturally subjected to significant stress during both tumorigenesis and anti-cancer therapy, there is growing evidence to suggest that modifications in the formation of SGs may serve as a protective mechanism for cancer cells against apoptosis[15]. Therefore, further investigation is required to determine the prognostic significance of SGs formation in HCC.
Throughout this investigation, a comprehensive analysis of SGs was performed in HCC. The gene set associated with SGs was obtained from the RNA granule database (rnagranuledb.lunenfeld.ca) and the research conducted by Youn et al[16]. Sequencing and relevant clinical data were obtained from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases for a cohort of 340 patients diagnosed with HCC. The objective was to develop prognostic models. The GEO database was utilized to obtain the datasets that were used for validating the expression of SG genes, specifically, GSE25097 and GSE36376. According to the aforementioned data sources, a comprehensive bioinformatics analysis was conducted. Using the SG genes in HCC, a risk scoring system HCC was developed and subsequently validated using the TCGA dataset. Furthermore, functional analysis and Gene Set Enrichment Analysis (GSEA) of SG genes investigated these genes’ prospective functions and pathways within HCC. These investigation outcomes show that signatures of seven SG genes can serve as independent predictors of overall survival (OS) in patients with HCC, and survival and prognosis analyses have been conducted for each expression of the hub gene.
MATERIALS AND METHODS
Data collection and pre-processing
This analysis was performed by downloading liver cancer data sets GSE25097 and GSE36376 from GEO official website (https://www.ncbi.nlm.nih.gov/geo/) database[17-26], and the datasets utilized in the study were derived from samples of Homo sapiens. The study employed the GPL10687 Rosetta/Merck Human RSTA Affymetrix 1.0 microarray and Custom CDF to analyze the GSE25097 dataset. This dataset consisted of 289 normal samples and 268 cases of HCC. The dataset GSE36376 is based on the platform GPL10558 Illumina HumanHT-12 V4.0 expression beadchip, and 0 normal samples and 433 HCC samples are included in the analysis.
Utilization of the UCSC Xena platform (https://xenabrowser.net/datapages/) allowed for the acquisition of TCGA dataset for this investigation. We downloaded TPM expression profile data, count data matrix, survival data, and clinical data of bulk-seq transcriptome sequencing of cancer tissues (371 cases) and neighboring tissues (50 cases) of HCC patients from UCSC Xena[27]. UCSC Xena has performed log2 normalization on its stored data to eliminate the dimension of gene expression data within samples. We used the annotation file (gencode.v22.annotation.gene.probeMap) provided by UCSC Xena to perform Gene Symbol re-annotation on the expression data to conduct an upcoming analysis. Download the clinical data of respective individuals through transcriptome sequencing, including gender, age, pathological condition, and survival prognosis. To achieve diverse analytical objectives, we decided to exclude patients with incomplete prognostic data in the context of endometrial cancer. We then integrated this data with GSE25097 and GSE36376 to obtain sequencing data and pertinent clinical information for 340 individuals with liver malignancy (Table 1). This was done to construct a prognosis model. The gene set pertaining to SGs utilized in this investigation was obtained from the RNA granule database (rnagranuledb.lunenfeld.ca) as well as from the study conducted by Youn et al[16]. SG genes with relevance score > 1 were extracted, and 241 genes were finally identified.
Least absolute shrinkage and selection operator-Cox regression analysis to screen prognostic genes
To assess the prognostic capacity of gene expression for normal and HCC groups, we extracted SG-related gene expression using a limma package based on the combined data set. Subsequently, the study employed univariate lasso, multivariate Cox, and Cox regression analyses to screen the prognostic genes and construct the prognostic model. Initially, the study used a univariate Cox proportional regression analysis to investigate the connection between differential gene expression and OS and retained genes with a P value of less than 0.05 (or 0.1). After that, The Least absolute shrinkage and selection operator (LASSO) algorithm was employed in the univariate Cox regression analysis in order to eliminate multicollinearity and determine which variables were significant. Multivariate Cox regression analysis was used to obtain more accurate independent prognostic factors, specifically hub genes. The final screening was conducted through stepwise regression. Ultimately, the algorithm for calculating risk scores was developed by integrating optimized gene expression data with the corresponding estimated Cox regression coefficients:
Where Ci exhibits the level of gene expression, Gi and is the coefficient related to the gene.
The specimens were categorized into two distinct groups, specifically the high- and low-risk groups, in accordance with the given risk score. The OS of the test set was analyzed through the utilization of a survival package, wherein both of log-rank test and Kaplan-Meier analysis were conducted. Survival and prognosis for each hub gene expression were analyzed. The study conducted an assessment of the reliability of survival prognostication through the application of time-dependent receiver operating characteristic (ROC) curves. The precision of prognosis or prediction was evaluated by computing the area under the curve (AUC) values using the time ROC R package. To explore differentially expressed genes (DEGs) across risk groups, we visualized differences in expression based on the Wilcoxon test and plotted boxplots.
Kyoto Encyclopedia of Genes and Genomes and Gene Ontology functional analysis
The Kyoto Encyclopedia of Genes and Genomes (KEGG)[28] database is a prevalent resource for the storage of data related to biological pathways, genomes, drugs, and diseases. KEGG[29] pathway enrichment of necroptosis differential genes was conducted utilizing R package clusterProfiler (version 4.2.0) to identify biological processes with significant enrichment, and the enrichment analysis was conducted and revealed a significance threshold of P value < 0.05. The present study performed a Gene Ontology (GO) enrichment analysis on the DEGs derived from the combined datasets of TCGA-Liver Hepatocellular Carcinoma (TCGA-LIHC), GSE25097, and GSE36376[30], which is a prevalent approach utilized in genes large-scale functional enrichment analyses across various categories and stages, the utilization of Fisher’s exact test for the computation of biological processes, respectively, (biological process), molecular function, and cellular component, the process of quantifying the differential genes contained within each GO entry was performed, and the study employed the hypergeometric distribution algorithm to assess the statistical significance level of the differential gene enrichment within every GO entry. The result of the calculation returns an enrichment significance P value, with lower values indicating more statistically significant. A P value less than 0.05 is reported as statistically significant. This article plays a part in visualizing it through bubble plots.
Gene Set Variation Analysis and GSEA enrichment assays
After screening DEGs among different risk groups, for exploring the pathway enrichment of differential genes, the present study conducted an enrichment analysis utilizing the Gene Set Variation Analysis (GSVA) approach on the set of (DEGs) in the combined datasets of TCGA-LIHC, GSE25097, and GSE36376 utilizing the GSVA package[31]. By downloading the gene set “c2.cp.kegg.v7.5.1.symbols.gmt” and setting the threshold as |log(fold change)| > 0.1 and P value < 0.05, we found pathways with significant enrichment differences among different subtypes. The GSVA technique is an unsupervised, non-parametric analytical approach. It is primarily employed to assess the outcomes of gene set enrichment analyses conducted on microarray and transcriptome data. The primary purpose of this technique is to assess the enrichment of distinct metabolic pathways across various samples. This is achieved by converting the gene expression matrix from one sample to another, resulting in a gene set expression matrix between samples.
Subsequently, GSEA has been conducted on all genes in the combined datasets of TCGA-LIHC, GSE25097, and GSE36376 using the “clusterprofiler” R package. GSEA is a statistical method employed to estimate the distribution pattern of genes belonging to a pre-defined gene set within a gene table that is ranked based on the degree of association with a phenotype to judge their significance to phenotype[32]. The gene set is denoted as “c2.cp.v7.5.1.symbols.gmt”[33] was acquired from the MSigDB database (v7.5.1) and carried out GSEA analysis on all genes respectively, and set P value < 0.05 was reported as statistically significant.
Protein-protein interaction
The STRING database[34] serves as a platform for identifying potential interactions between established and projected proteins. Based on the combined datasets of TCGA-LIHC, GSE25097, and GSE36376, we established the networks of protein-protein interaction (PPI) for DEGs utilizing the STRING database. The STRING database (version 11.5) was utilized to upload the gene list, and the reliability was set to 0.4; PPI table data was subsequently purchased into Cytoscape (version 3.9.1) software for constructing PPI networks for visualization. Finally, the utilization of CytoHubba plug-in was employed to examine the hub genes that are present within the PPI network, and the top ten core genes were identified and chosen according to MCC score (Maximum clique centrality)[35]. Utilizing the Human Protein Atlas, database facilitated the visualization of core gene expression in various tissues.
Building a prediction nomogram
Based on the TCGA-LIHC, GSE25097, and GSE36376 combined datasets, we incorporated risk scores and clinicopathological characteristics into the model, followed by the application of multivariate Cox regression for assessing the risk scores impact and clinicopathological features on prognosis. The RMS package (version:5.1-4, http://cran.r-project.org/web/packages/rms/index.html) was then utilized for the production of nomograms and calibration plots that encompassed clinically relevant characteristics. The evaluation of calibration was conducted by means of a process that involved the mapping of anticipated probability derived from the nomogram to the actual probability of occurrence, considering the 45° line representing the best predictive value. The utilization of decision curve analysis (DCA) allowed for the evaluation of risk score clinical values in addition to the clinical pathological characteristics exhibited by the model with respect to the potential advantages for the patient. The larger the area under the curve, the more significant clinical benefit the patient would have.
Statistical method
The present investigation’s data processing and analysis were conducted using R software (Version 4.1.0). The continuous variables are presented as mean ± SD. The comparison of progression-free survival within the two groups was carried out through the utilization of Kaplan-Meier analysis in combination with the log-rank test. χ2 or Fisher’s exact tests were utilized to assess and examine the statistically significant level between two sets of categorical variables. If not specified, the results were calculated and obtained by Spearman correlation analysis between the correlation coefficients of different molecules, and all statistical P values utilized in the analysis were two-sided, with P < 0.05, which was deemed statistically significant.
RESULTS
Project flow chart
In this study, the liver cancer data were downloaded from the GEO official website database and TCGA database for analysis. The project flow chart is shown in Figure 1.
Figure 1 Project flow chart.
TCGA-LIHC: The Cancer Genome Atlas-Liver Hepatocellular Carcinoma; LASSO: Least absolute shrinkage and selection operator; PPI: Protein-protein interaction; GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; GSEA: Gene Set Enrichment Analysis; GSVA: Gene Set Variation Analysis; ROC: Receiver operating characteristic curve; DCA: Decision curve analysis.
Risk model construction
Following the extraction of SG gene expression in HCC, aiming to predict the clinical risk and survival outcome of samples through the difference of SG gene expression and function, based on the combined dataset of TCGA-LIHC, GSE25097, and GSE36376, we used LASSO model method to determine the optimal hub gene number (Figure 2A) and correspondence coefficient (Figure 2B), screened the best hub genes according to the ratio of 5:5, and divided the HCC samples into train group and test group. Finally, the genes for constructing risk scores were determined. The corresponding correlation coefficients were calculated based on multi-factor COX regression (Table 2) for calculating the risk score according to the hub genes expression level (Supplementary Table 1), and training specimens were separated into two different groups, which relied on the HCC samples median risk score (Figure 2C), and test group samples (Figure 2D) have been separated into two distinct groups according to their level of risk; namely, the high-risk group (high risk) and the low-risk group (low risk), and hub genes expression in train group specimens (Figure 2E) and test group samples (Figure 2F) within the high- and low-risk groups was visualized through the use of heat map. The red color indicates the upregulation of gene expression level within the sample, and the blue color represents the under-expression of gene expression level within the sample risk score = DDX1* 1.109698909 + DKC1* 0.624264345 + BICC1* - 0.489537897 + HNRNPUL1* - 2.330604694 + CNOT6* 1.986647774 + DYRK3* 0.584581122 + CCDC124* 0.635540329.
Figure 2 Risk grouping construction based on The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Least absolute shrinkage and selection operator is a gradient descent method for determining hub genes; B: Number of hub genes and their corresponding coefficients; C: Train group samples were classified into high- and low-risk groups based on the risk scores. Red indicates high-risk group samples, and blue indicates low-risk group samples; D: Test group samples have been classified into high-risk and low-risk groups based on the risk scores. Red indicates high-risk group samples, and blue indicates low-risk group samples; E: The hub genes expression within the high- and low-risk groups of the train group. Red indicates up-regulated expression, blue indicates under-expression of the expression, and white indicates zero expression; F: The hub genes expression within high- and low-risk groups of the test group. Red indicates up-regulated expression, blue indicates under-expression of the expression, and white indicates zero expression.
Table 2 Hubgenes screened by Least absolute shrinkage and selection operator with coefficients.
We then performed survival analysis on specimens from high- and low-risk groups within the combined dataset of TCGA-LIHC, GSE25097, and GSE36376. Survival analysis revealed that the samples of the low-risk group within the training group (Figure 3A) (P < 0.001) and the test group (Figure 4A) (P = 0.022) had longer survival times. Based on this, a survival analysis of all hub genes within the train group and test group was performed, respectively, and it was revealed that each hub gene expression within the train group and test group had significant differences in HCC survival. CCDC124 (P < 0.001) (Figure 3B), DDX1 (P < 0.001) (Figure 3C), DKC1 (P < 0.001) (Figure 3D), HNRNPUL1 (P = 0.043) (Figure 3E), CNOT6 (P < 0.001) (Figure 3F), BICC1 (P = 0.001) (Figure 3G), DYRK3 (P = 0.004) (Figure 3H) in train group; in the test group, CCDC124 (P < 0.001) (Figure 4B), DDX1 (P < 0.001) (Figure 4C), DKC1 (P < 0.001) (Figure 4D), HNRNPUL1 (P = 0.002) (Figure 4E), CNOT6 (P < 0.001) (Figure 4F), BICC1 (P = 0.002) (Figure 4G), DYRK3 (P = 0.006) (Figure 4H). Then the differential genes were screened among high and low-risk groups in accordance with the Wilcoxon rank sum test (Supplementary Table 2).
Figure 3 Survival analysis of high- and low-risk groups and hub genes in train group specimens according to The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Survival analysis of risk scores for train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; B: Survival analysis of CCDC124 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; C: Survival analysis of DDX1 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; D: Survival analysis of DKC1 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; E: Survival analysis of HNRNPUL1 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; F: Survival analysis of CNOT6 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; G: Survival analysis of BICC1 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples; H: Survival analysis of DYRK3 in train group samples. Red is indicative of high-risk group samples, and blue is indicative of low-risk group samples.
Figure 4 Hub genes survival analysis within the test group.
A: Survival analysis of risk scores for test group samples. Red represents high-risk group samples, and blue indicates low-risk group samples; B: Survival analysis of CCDC124 samples in the test group. Red indicates high-risk group samples, and blue represents low-risk group samples; C: Survival analysis of DDX1 in test group samples. Red indicates high-risk group samples, and blue indicates low-risk group samples; D: Survival analysis of DKC1 in test group samples. Red represents high-risk group samples, and blue indicates low-risk group samples; E: Survival analysis of HNRNPUL1 in the test group. Red represents high-risk group samples, and blue indicates low-risk group samples; F: Survival analysis of CNOT6 in test group samples. Red indicates high-risk group samples, and blue indicates low-risk group samples; G: Survival analysis of BICC1 in test group samples. Red indicates high-risk group samples, and blue indicates low-risk group samples; H: Survival analysis of DYRK3 in test group samples. Red indicates high-risk group samples, and blue indicates low-risk group samples.
Functional analysis
The present study conducted functional typing of DEGs among the high-risk and low-risk groups within the combined TCGA-LIHC, GSE25097, and GSE36376 datasets. In order to determine biological processes that are significantly enriched, the differential gene expression analysis underwent GO and KEGG enrichment analysis via the clusterProfiler R package (Tables 3 and 4). Each term in GO enrichment results is sorted in ascending order according to P value, i.e., the more significant the enrichment, the higher. We selected the first 12 GO terms for circle diagram display (Figure 5A) and then selected the first 4 terms of the three functional categories to visualize with a bar chart (Figure 5B). It can be seen that in GO analysis, significantly different necroptosis genes (P < 0.05) have primarily participated in a collection of biological activities like the regulation and translation of the mRNA metabolic process, RNA catabolic process, and mRNA catabolic process (Figure 5D), and also include cell components such as cytoplasmic SG, cytoplasmic RNP granule, RNP granule and p-body (Figure 5E), and enriched molecular functions such as nucleic acid binding, translation regulator activity, translation factor activity, translation initiation factor activity, and RNA binding (Figure 5F). As shown in the findings of KEGG enrichment analysis, the genes whose expression levels were found to be differential have been enriched in the RNA degradation process, amyotrophic lateral sclerosis, spliceosome and nucleocytoplasmic transport pathways (Figure 5C), and the connection among gene expression and pathway enrichment was shown by string diagram for the first five pathways (Figure 5G).
Figure 5 Gene Ontology analysis and Kyoto Encyclopedia of Genes and Genomes analysis of differentially expressed genes between high- and low-risk groups in The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: The circle diagram visualization Gene Ontology (GO) enrichment analysis outcomes among the high- and low-risk groups, the outer circle is GO terms, red dots indicate genes overexpressed genes, blue dots indicate under-expressed genes, quadrilateral colors indicate the z-score of GO terms, blue indicates the z-score is negative, and the corresponding GO terms would probably exhibit inhibition, and red indicates z-score is positive, and it is more probable for the activation of the term to take place in the corresponding GO terms; B: Dot plot of GO enrichment analysis for differentially expressed genes; C: The abscissa represents the gene ratio. The ordinate represents the pathway name, the node size indicates the genes enriched number included within the pathway, and the node color is indicative of the P value; D: Biological processes function enrichment chord diagram, node color represents gene expression level. Red indicates the gene overexpressed gene, and blue indicates the under-expressed gene; E: Cellular component function enrichment chord diagram, node color represents gene expression level. Red indicates the gene overexpressed gene, and blue indicates the under-expressed gene; F: Molecular function enrichment chord diagram, node color represents gene expression level. Red indicates the gene overexpressed gene, and blue indicates the under-expressed gene; G: Kyoto Encyclopedia of Genes and Genomes function enrichment chord diagram, node color represents gene expression level. Red indicates the gene overexpressed gene, and blue indicates the under-expressed gene. FC: Fold change.
We then performed GSVA and GSEA analyses using the GSVA package and clusterProfiler R package for all genes in the TCGA-LIHC, GSE25097, and GSE36376 combined datasets (Tables 5 and 6). GSVA analyses showed that the low-risk group exhibited primarily enriched results and up-regulated in vasopressin-regulated water reabsorption, cell cycle, progesterone-mediated oocyte maturation, basal transcription factors, and RNA degradation pathways compared to the high-risk group (Figure 6); It is enriched and down-regulated in maturity-onset diabetes of the Young, calcium signaling pathway, neuroactive ligand-receptor interaction, glycine serine and threonine metabolism and peroxisome proliferator-activated receptor (PPAR) signaling pathway. GSEA analysis suggests low risk versus high risk in fatty acid metabolism (Figure 7A), retinal metabolism (Figure 7B), drug metabolism cytochrome P450 (Figure 7C), PPAR signaling pathway (Figure 7D), steroid hormone biosynthesis (Figure 7E), threonine and glycine serine metabolism (Figure 7F), cell cycle (Figure 7G), butanoate metabolism (Figure 7H), complement and coagulation cascades (Figure 7I) and other pathways have significant enrichment differences (P < 0.05).
Figure 6 Gene Set Variation Analysis and Gene Set Enrichment Analysis of differential genes among high- and low-risk groups within The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
The main biological functions and pathway heat maps obtained from Gene Set Variation Analysis are displayed. Blue indicates low risk, and yellow indicates high risk. Red represents overexpression in this pathway, and blue indicates under-expression in this pathway. KEGG: Kyoto Encyclopedia of Genes and Genomes.
Figure 7 Gene Set Enrichment Analysis of differentially expressed genes between high- and low-risk groups in The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Gene Set Enrichment Analysis (GSEA) analysis illustrated that low risk was significantly up-regulated within the KEGG_FATTY_ACID_METABOLISM pathway compared to high risk; B: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_RETINOL_METABOLISM pathway compared to high risk; C: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_DRUG_METABOLISM_CYTOCHROME_P450 pathway compared to high risk; D: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_PPAR_SIGNALING_PATHWAY pathway compared to high risk; E: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_STEROID_HORMONE_BIOSYNTHESIS pathway compared to high risk; F: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_GLYCINE_SERINE_AND_THREONINE_METABOLISM pathway compared to high risk; G: GSEA analysis illustrated that low risk was significantly down-regulated within the KEGG_CELL_CYCLE pathway compared to high risk; H: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_BUTANOATE_METABOLISM pathway compared to high risk; I: GSEA analysis illustrated that low risk was significantly up-regulated within the KEGG_COMPLEMENT_AND_COAGULATION_CASCADES pathway compared to high risk. KEGG: Kyoto Encyclopedia of Genes and Genomes.
A SG gene-based PPI network was established, utilizing differential expression data among the high-risk and low-risk groups within the combined dataset of TCGA-LIHC, GSE25097, and GSE36376 through the STRING website, and evaluated the obtained data and visualized the PPI network utilizing Cytoscape software, where red indicates overexpressed genes and blue indicates underexpressed genes (Figure 8A); subsequently, a set of ten genes were chosen as the core genes (EIF3D, EIF4G1, EIF4A2, EIF3B, EIF3A, EIF3E, EIF4B, EIF4E, DDX3X, and EIF3H) using cytoHubba plug-in analysis of MCC scores (Figure 8B).
Figure 8 Protein-protein interaction analysis.
A: Protein-protein interaction analysis was conducted utilizing the STRING database for kinetokines-related genes, and the visualization of interaction relationship was carried out, where red indicates up-regulated genes and blue indicates downregulated genes; B: The resulting top 10 hub genes were analyzed utilizing the CytoHubba plug-in.
Hub gene correlation and prognosis analysis
For evaluating the influence of hub genes on HCC prognosis, we applied univariate Cox analysis to hub genes in TCGA-LIHC, GSE25097, and GSE36376 combined datasets and found that hub genes had a significant influence on HCC (P < 0.05) (Figure 9A). Concurrently, the calculation of hazard ratio value for each hub gene was performed to determine whether the gene was a protective or risk factor (Table 7). We then calculated and visualized the association among the hub genes (Figure 9B), and all seven of them showed positive correlations. The ROC curve for the prognostic model utilizing hub genes was plotted through the implementation of time ROC package for evaluating its performance and calculated the model’s AUC values at 1, 3, and 5 years within the training group (Figure 9C) and the test group (Figure 9D). The 1-, 3-, and 5-year survival rate AUC of train group were 0.886, 0.806, and 0.755, respectively (Figure 9C). The 1-, 3-, and 5-year survival AUCs in the Test group were 0.730, 0.694, and 0.683, respectively (Figure 9D).
Figure 9 Hub gene association and prognosis analysis relied on The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Univariate Cox analysis of Hub genes in hepatocellular carcinoma (HCC). The left side of the dotted line represents hazard ratio (HR) < 1, revealing that this gene expression serves as a protective factor for the prognosis of HCC; the right side of the dotted line represents HR > 1, suggesting that this gene expression is a predictive risk factor for HCC; B: Visualization of interactions between Hub-genes. The red line indicates a positive association between the two different genes; the size of dot indicates the P value of the gene’s effect on the prognosis of HCC samples, and the orange dot represents that the gene is a risk factor for HCC; C: Receiver operating characteristic (ROC) curve. Green represents 1-year survival, blue represents 3-year survival, and red represents 5-year survival; D: ROC curve of test group. Green represents 1-year survival, blue represents 3-year survival, and red represents 5-year survival. AUC: Area under the curve.
Prognostic model construction
A prognostic model was constructed by incorporating risk score, age, T stage, and N stage, and based on the combined dataset of TCGA-LIHC, GSE25097, and GSE36376, multivariate Cox regression revealed that risk score (P < 0.001), grade (P < 0.001), age (P = 0.022), and T stage (P < 0.001) were significantly correlated with HCC prognosis (Figure 10A). The risk stratification curve demonstrates that the prognostic model can well anticipate the survival time of HCC samples in different risk groups (Figure 10B), while the calibration curve shows that the overfitting control of the model is reasonable and the preparation is high (Figure 10C). We then visualized the prognostic model via nomograms, based on which 1-, 3-, and 5-year survival rates for HCC could be anticipated (Figure 11A). The DCA analysis showed that nomograms based on prognostic models were associated with a more significant clinical benefit in HCC patients at 1-year (Figure 11B), 3-year (Figure 11C), and 5-year (Figure 11D) survival.
Figure 10 Multivariate Cox analysis and model performance of prognostic factors based on The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Visualization of the correlation between risk scores and clinical characteristics and hepatocellular carcinoma (HCC) prognosis, aP ≤ 0.05, bP ≤ 0.01, cP ≤ 0.001; B: The prognostic model classified the HCC sample into high-risk and low-risk groups, and there was a significant variance in survival among the high- and low-risk groups. Red is indicative of the high-risk group, and blue is indicative of the low-risk group; C: Calibration curve of HCC clinical prognosis model. Green represents 1-year survival, blue represents 3-year survival, and red represents 5-year survival. OS: Overall survival.
Figure 11 Prognostic model nomograms and decision curve analysis curves based on The Cancer Genome Atlas-Liver Hepatocellular Carcinoma, GSE25097, and GSE36376 combined datasets.
A: Clinical prognostic model nomogram of hepatocellular carcinoma (HCC). aP ≤ 0.05, cP ≤ 0.001; B: Decision curve analysis (DCA) curve for 1-year survival. The area under the curve represents the possible prognostic benefit for HCC patients based on this factor; C: DCA curve for 3-year survival. The area under the curve represents the possible prognostic benefit for HCC patients based on this factor; D: DCA curve for 5-year survival. The area under the curve represents the possible prognostic benefit for HCC patients based on this factor.
DISCUSSION
The prevalence of liver cancer has been observed to increase recently. HCC is the most commonly occurring primary liver cancer disorder. Since HCC does not respond to radiotherapy as well as chemotherapy, and there is no effective treatment for unresectable HCC, the mortality of HCC currently ranks second globally, and the 5-year survival rate is lower than 15%[36,37]. Recently, several studies have shown that SGs’ biogenesis and stability are related to cancer progression[14]. Cancer cells frequently encounter hypoxic, nutrient-deprived, and hyperosmotic environments to obtain their heightened metabolic requirements for proliferation[38]. Therefore, the tumor microenvironment is often full of various distresses, including high concentrations of reactive oxygen species and hypoglycemia, almost all of which may closely induce the formation of SGs[11]. SG is involved in tumorigenesis and cancer metastasis through tumor-related signaling pathways and other mechanisms. In addition, some chemotherapeutic agents were shown to promote SGs[39]. Hence, SGs’ role in cancer has garnered significant attention. Upregulation of SG components has been observed in HCC[39]. Nevertheless, additional research is required to discover the prognostic significance of SGs in HCC.
Throughout this investigation, the sequencing and relevant clinical data of individuals diagnosed with HCC were acquired from the combined datasets of TCGA-LIHC, GSE25097, and GSE36376. SG genes with relevance score > 1 were extracted from the RNA granule database (rnagranuledb.lunenfeld.ca), and the study of Youn et al[16], and a total of 241 genes were finally detected. Then the optimal number of hub genes was identified by the LASSO model, and the optimal hub genes were screened according to the ratio of 5:5. HCC samples were separated into train and test groups. Therefore, the median risk score is computed according to the hub genes expression, and the train samples and test samples are separated into high- (high risk) and low-risk groups (low risk). Survival analysis revealed that the low-risk group within the train and test groups had a higher OS rate and longer survival time. Between them, the7 hub genes expression levels (CCDC124, DDX1, DKC1, HNRNPUL1, CNOT6, BICC1, DYRK3) in the train group and test group had significant variations in survival time and OS of HCC (P < 0.05). Functional analysis has been conducted on these seven differential genes. In GO analysis, significant differential genes (P < 0.05) were primarily contributed within various biological processes, including modulation of mRNA metabolic, mRNA catabolic, RNA catabolic processes, and translation regulation. Also, they contained cell components that were rich in RNP, cytoplasmic RNP, cytoplasmic SGs, and p-body. According to the KEGG enrichment analysis, genes that exhibited differential expression were rich in the RNA degradation process, amyotrophic lateral sclerosis, spliceosome, and nucleocytoplasmic transport pathways. GSEA analysis revealed that low risk was significantly enriched in retinol metabolism, fatty acid metabolism, drug metabolism cytochrome P450, steroid hormone biosynthesis, PPAR signaling pathway, serine, glycine and threonine metabolism, cell cycle, butanoate metabolism, complement, and coagulation cascades compared to high risk (P < 0.05). We further analyzed the PPI of kinetogranin-related genes using the STRING database and revealed the top ten up-regulated core genes (EIF3D, EIF4G1, EIF4A2, EIF3B, EIF3A, EIF3E, EIF4B, EIF4E, DDX3X, EIF3H). Univariate cox analysis showed that seven hub genes (DDX1, DKC1, BICC1, HNRNPUL1, CNOT6, DYRK3, CCDC124) were risk factors for HCC prognosis. Through the utilization of risk score, multivariate Cox regression was identified (P < 0.001), grade (P < 0.001), age (P = 0.022), and T stage (P < 0.001) were significantly connected with HCC prognosis. Through risk stratification curve analysis, we confirmed that the prognostic model can well anticipate the HCC samples’ survival time within different risk groups. DCA analysis demonstrated that the prognostic model can make HCC individuals have higher clinical benefits in 1-, 3- and 5-year survival rates. Our findings suggest that the differential expression of SG-related genes is significantly connected to the OS rate, survival time, and prognosis of HCC.
SGs are induced by various stress conditions (heat, oxidative stress, and hypoxia). In cancer cells, the formation of SGs is thought to protect them from apoptosis and induce resistance to anti-cancer drugs or radiotherapy, making SGs a potential target for cancer therapy[40]. Most current understanding of stress particles is still in the molecular and cellular biological stages, and their practical impact on the human clinical environment is still insufficient. This study further confirmed that seven SG genes (DDX1, DKC1, BICC1, HNRNPUL1, CNOT6, DYRK3, and CCDC124) were significantly correlated with survival time of individuals with HCC and the OS rate and were also risk factors affecting the prognosis of HCC, providing a more theoretical basis for SGs as an innovative biomarker and prospective therapeutic target for the management of HCC. DDX1, DYRK3, and CCDC124 have been reported to be overexpressed in HCC[41-43], but DKC1, BICC1, HNRNPUL1, and CNOT6 are rarely reported in HCC.
DDX1 belongs to the DEAD-box RNA helicase family and contributes to various biological processes, including mRNA translation, tRNA splicing, rRNA processing, miRNA maturation, and DNA double-strand break (DSB) repair[44,45]. DDX1 has been found to promote the development of cancers, including neuroblastoma, retinoblastoma, testicular cancer, colorectal malignancy, breast malignancy, and liver cancer[41,46-48]. The study by Yuan et al[41] suggested that DDX1 is a poor prognostic factor of liver cancer, which is consistent with this study. In addition, the DDX1 promoter methylation level within the HCC tissues exhibited reduced levels than in normal tissues. The positive association between DDX1 expression and infiltration of immune cells (including dendritic cells, macrophages, B cells, and T cells) is primarily involved in the cell cycle and RNA editing[41].
Kan et al[49] showed that the DKC1 gene performs a vital function in the progression of cells in colorectal cancer, and a higher DKC1 expression level induces colorectal cancer progression by increasing ribosomal protein expression in a pseudouridine synthase activity-dependent manner. Current studies have shown that BICC1 can regulate biological processes, including apoptosis and proliferation, and has a poor association, prognosis for oral cancer and gastric cancer, and immune infiltration[50-52].
HNRNPUL stimulates the end excision of DNA, induces DNA DSB repair via homologous recombination, and participates in DNA damage response. It has been discovered that higher HNRNPUL1 expression levels are associated with an increased likelihood of future occurrences and are an independent risk factor for disease-free survival in esophageal squamous cell carcinoma individuals administrating platinum-based chemotherapy following the surgery[53].
CNOT6 has been considered to be up-regulated within acute lymphoblastic leukemia, acute myeloid leukemia, androgen-independent prostate cancer, and lung cancer cells, and expression of CNOT6 affects DNA mismatch repair[54]. The expression of DYRK1A and DYRK3 exhibits a correlation with immune-infiltrating cells within the tumor microenvironment and is overexpressed in MSI subtypes, which can serve as biomarkers for immunotherapy[42].
CCDC124 is a cell division-associated RBP significantly up-regulated in esophageal, adrenal, endometrial, liver, ovarian, thyroid, and bladder cancers[43]. In the present investigation, differential expression of the seven SG genes in HCC is found to affect the OS rate, survival time, and prognosis of HCC patients. The SG becomes a biomarker for predicting the stage of progression and the therapeutic effect of tumors, and the targeted SG can serve as a new method for tumor treatment. However, additional research is necessary to understand the specific mechanism fully.
Furthermore, the differential expression of SG-related genes in HCC was found, which was related to RNA degradation, nucleic acid transport, and splicing pathways, as well as fatty acid, retinol, cytochrome P450, PPAR, sterol synthesis, amino acid metabolism, and other pathways. In GO analysis, the significantly different necroptosis genes primarily participated in mRNA metabolic process regulation, translation regulation, RNA catabolism process, mRNA catabolism process, and other different biological processes, as well as cellular components such as RNP particles, cytoplasmic RNP particles, cytoplasmic stress particles, and p-bodies. Moreover, the findings of KEGG enrichment analysis indicate that the genes exhibiting differential expression are significantly enriched in the process of RNA degradation, amyotrophic lateral sclerosis, spliceosome, and nucleoplasmic transport pathways. Further GSVA analysis showed that according to the low-risk group, the high-risk group primarily enriched and up-regulated the vasopressin-regulated water reabsorption, cell cycle, progesterone-mediated oocyte maturation, basal transcription factors, and RNA degradation pathways. It is enriched and downregulated in young-onset adult-onset diabetes, calcium signaling pathway, neuroactive ligand-receptor interaction, glycine, serine and threonine metabolism, and PPAR signaling pathway. GSEA analysis revealed that there were significant enrichment variations between the high- and low-risk groups throughout drug metabolism, retinal metabolism, fatty acid metabolism, cytochrome P450, PPAR signaling pathway, steroid hormone biosynthesis, serine, glycine and threonine metabolism, cell cycle, butyrate metabolism, complement, and coagulation cascade.
Although our present investigation offers novel perspectives on the correlation between stress particles and prognostic significance in patients with HCC, it is crucial to acknowledge the limitations that must be taken into account. At first, this investigation involved only one dataset, which may have led to selection bias. Second, due to the use of online databases as the primary source of data in this study, certain critical clinical data, including specific chemotherapy treatments administered to the patients, could not be obtained. Third, additional research and thorough in vivo and in vitro experimental verification are crucial to clarify the biological roles and fundamental mechanisms of SGs in HCC.
CONCLUSION
In conclusion, from the TCGA and GSE datasets, we found that seven SG genes (DDX1, DKC1, BICC1, HNRNPUL1, CNOT6, DYRK3, and CCDC124) exhibited differential expression within HCC. GO and GSEA analysis revealed that these SGs may be involved in RNA degradation, nucleic acid transport, splicing pathways, as well as fatty acid, retinol, cytochrome P450, PPAR, sterol synthesis, and amino acid metabolism. Nomogram models with good prognostic accuracy were constructed for 1-, 3-, and 5-year OS predictions. These may contribute novel ideas to the study of HCC. Differential expression of the seven SG genes in HCC is found to affect the OS rate, survival time, and prognosis of HCC patients. The SG becomes a biomarker for predicting the stage of progression and the therapeutic effect of tumors, and the targeted SG can serve as a new method for tumor treatment. Although, the data mentioned above were obtained through bioinformatics analysis and required confirmation from a significant number of prospective study samples.
ACKNOWLEDGEMENTS
We acknowledge The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) database for providing their platforms and contributors for uploading their meaningful datasets. TCGA and GEO belong to public databases. The patients involved in the database have obtained ethical approval. Users can download relevant data for free for research and publish relevant articles. Our study is based on an open source data, so there are no ethical issues and other conflicts of interest.
Santopaolo F, Lenci I, Milana M, Manzia TM, Baiocchi L. Liver transplantation for hepatocellular carcinoma: Where do we stand?World J Gastroenterol. 2019;25:2591-2602.
[PubMed] [DOI] [Full Text]
Ogunwobi OO, Harricharran T, Huaman J, Galuza A, Odumuwagun O, Tan Y, Ma GX, Nguyen MT. Mechanisms of hepatocellular carcinoma progression.World J Gastroenterol. 2019;25:2279-2293.
[PubMed] [DOI] [Full Text]
Lamb JR, Zhang C, Xie T, Wang K, Zhang B, Hao K, Chudin E, Fraser HB, Millstein J, Ferguson M, Suver C, Ivanovska I, Scott M, Philippar U, Bansal D, Zhang Z, Burchard J, Smith R, Greenawalt D, Cleary M, Derry J, Loboda A, Watters J, Poon RT, Fan ST, Yeung C, Lee NP, Guinney J, Molony C, Emilsson V, Buser-Doepner C, Zhu J, Friend S, Mao M, Shaw PM, Dai H, Luk JM, Schadt EE. Predictive genes in adjacent normal tissue are preferentially altered by sCNV during tumorigenesis in liver cancer and may rate limiting.PLoS One. 2011;6:e20090.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 70][Cited by in RCA: 62][Article Influence: 4.1][Reference Citation Analysis (0)]
Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, Mulawadi FH, Wong KF, Liu AM, Poon RT, Fan ST, Chan KL, Gong Z, Hu Y, Lin Z, Wang G, Zhang Q, Barber TD, Chou WC, Aggarwal A, Hao K, Zhou W, Zhang C, Hardwick J, Buser C, Xu J, Kan Z, Dai H, Mao M, Reinhard C, Wang J, Luk JM. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma.Nat Genet. 2012;44:765-769.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 803][Cited by in RCA: 753][Article Influence: 53.8][Reference Citation Analysis (4)]
Open-Access: This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Provenance and peer review: Unsolicited article; Externally peer reviewed.