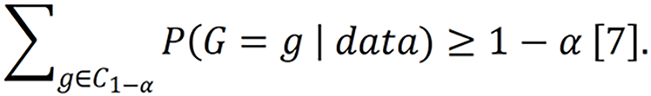Evangelou K, Angeli P, Polydorou A, Petropoulou T. Single-cell deoxyribonucleic acid typing for forensic mixtures and trace evidence: Opportunities, validation requirements, and reporting limits. World J Biol Chem 2026; 17(2): 121467 [PMID: 42273528 DOI: 10.4331/wjbc.v17.i2.121467]
Corresponding Author of This Article
Kyriacos Evangelou, MD, Department of Minimally Invasive Colon and Rectal Surgery, The Euroclinic Hospital of Athens, 7-9 Athanasiadou & D. Soutsou Street, Athens 11521, Greece. evangeloukyriacos@gmail.com
Research Domain of This Article
Medicine, Legal
Article-Type of This Article
review-article
Open-Access Policy of This Article
This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Kyriacos Evangelou, Andreas Polydorou, Thalia Petropoulou, Department of Minimally Invasive Colon and Rectal Surgery, The Euroclinic Hospital of Athens, Athens 11521, Greece
Kyriacos Evangelou, Andreas Polydorou, Thalia Petropoulou, Second Department of General Surgery, Aretaieion University Hospital, Athens 11528, Greece
Paraskevi Angeli, Department of Biology, University of Cyprus, Nicosia 1678, Cyprus
Paraskevi Angeli, Department of Neurogenetics, Cyprus Institute of Neurology and Genetics, Nicosia 1683, Cyprus
Author contributions: Evangelou K, Angeli P, Polydorou A, and Petropoulou T contributed to manuscript writing and editing; Polydorou A and Petropoulou T contributed to data collection; Evangelou K and Angeli P contributed to data analysis; Polydorou A and Petropoulou T contributed to conceptualization and supervision; and all authors have read and approved the final manuscript.
Conflict-of-interest statement: All authors declare that they have no conflict of interest to disclose.
Corresponding author: Kyriacos Evangelou, MD, Department of Minimally Invasive Colon and Rectal Surgery, The Euroclinic Hospital of Athens, 7-9 Athanasiadou & D. Soutsou Street, Athens 11521, Greece. evangeloukyriacos@gmail.com
Received: March 26, 2026 Revised: April 24, 2026 Accepted: May 9, 2026 Published online: June 5, 2026 Processing time: 71 Days and 14.3 Hours
Abstract
Forensic deoxyribonucleic acid (DNA) interpretation is limited less by genotyping technology than by the biochemical and inferential effects of mixed, low-template, and environmentally complex traces. Single-cell and single-molecule strategies, including the United Kingdom Research and Innovation-funded single-cell and single-molecule analysis for DNA identification (SCAnDi) program, aim to preserve cellular resolution. They allow investigators to isolate and type individual cells or defined small-cell pools before heterogeneous evidence is converted into a bulk lysate. In selected validation settings, this approach has yielded near-complete diploid short tandem repeat (STR) profiles from small pools, credible genotype sets tightly concentrated on the true genotype across high-order mixtures, and improved access to donor-specific profiles from sexual assault and other complex samples. However, these studies also show important limits. Many operationally successful “single-cell” workflows are, in practice, single-cell-plus-consensus or few-cell workflows. Stochastic effects remain intrinsic, and cell capture itself becomes a probabilistic sampling step. This opinion review explicitly adopts an evaluative stance: It synthesizes recent validation studies and guidance documents to identify the performance thresholds and reporting boundaries that should be met before targeted forensic deployment of SCAnDi-like workflows. We argue that casework entry should require measured cell-recovery probabilities, phenotype-misclassification rates, locus- and cell-type-specific dropout and stutter models, quantified contamination and drop-in rates, validated minimum cell counts for consensus generation, and explicit database-upload criteria. It should also require strict separation between sub-source reporting and activity-level propositions. If those conditions are met, single-cell typing can complement, rather than replace, bulk STR analysis and probabilistic genotyping in a narrow but important set of high-value forensic scenarios.
Core Tip: Single-cell forensic deoxyribonucleic acid typing has moved beyond speculation, but present evidence supports targeted use only in validated niches. Its forensic value lies in reducing mixture complexity before computation, preserving cell-of-origin information, and rescuing contributor profiles that bulk analysis may not resolve. Its forensic risk lies in selective cell capture, single-template stochasticity, contamination at near-single-copy sensitivity, and the temptation to over-read cellular context as activity-level proof. The appropriate translational question is therefore not whether single-cell short tandem repeat typing works in principle, but which minimum validation thresholds and reporting limits are necessary before it enters accredited casework.
Citation: Evangelou K, Angeli P, Polydorou A, Petropoulou T. Single-cell deoxyribonucleic acid typing for forensic mixtures and trace evidence: Opportunities, validation requirements, and reporting limits. World J Biol Chem 2026; 17(2): 121467
Mixed deoxyribonucleic acid (DNA) profiles are a routine biochemical consequence of lysing heterogeneous cellular material into a single extract, not a rare artifact of poor technique[1,2]. In the single-cell and single-molecule analysis for DNA identification (SCAnDi) grant abstract, up to 45% of forensic samples in the United Kingdom criminal justice system are described as containing DNA from two or more individuals, often limiting database searching, whereas a nationwide Danish casework study reported that approximately two-thirds of reported forensic profiles from 2016 to 2024 were mixtures[3,4]. The same Danish study showed that lowering the polymerase chain reaction (PCR)-capillary electrophoresis input threshold from 200 pg to 100 pg increased the reported-profile rate from 50% to 59%. This illustrates the central modern trade-off: Greater analytical sensitivity improves yield, but it also increases the fraction of profiles dominated by stochastic effects and difficult interpretation[4,5].
Single-cell strategies are attractive because they seek to intervene before this inferential bottleneck forms, by keeping evidence cellular long enough to image, classify, isolate, and type individual cells or defined small-cell pools rather than immediately creating a pooled DNA population[2,3]. In the present opinion review, we use “single-cell” as an umbrella term unless otherwise specified. We recognize, however, that much of the strongest forensic evidence concerns true single cells interpreted through replication or consensus, or low-cell-number pools that are better described as cell-resolved than as literal one-cell complete genotyping[6-9]. Our purpose is therefore not to celebrate technical feasibility alone, but to identify the practical validation thresholds, database-compatibility conditions, and reporting limits that should be satisfied before targeted implementation in accredited forensic casework[3,6,10-12].
WHERE CURRENT MIXTURE INTERPRETATION SUCCEEDS AND WHERE IT STRUCTURALLY SATURATES
Probabilistic genotyping (PG) has substantially improved forensic mixture interpretation by modeling dropout, drop-in, stutter, peak-height behavior, contributor number, and competing propositions within a likelihood ratio (LR) framework rather than a binary inclusion/exclusion scheme[5,13]. This literature also includes developmental validation, internal validation, inter-system review, case-study comparison, and published critique of major PG platforms[14-18]. Contemporary guidance now emphasizes that laboratories must validate the intended factor space of application, define procedures for assigning the number of contributors, document which profile components are suitable for comparison, and communicate the proposition level addressed by the LR[19-22]. This progress matters for SCAnDi because cell-resolved electropherograms are not exempt from probability-based reasoning: They remain vulnerable to elevated stutter, imbalance, sporadic allele loss, and occasional contamination, and therefore still require validated models and transparent assumptions[6-8].
The structural saturation point of bulk short tandem repeat (STR) interpretation is reached when contributor number is high, allele sharing is extensive, contributor ratios are very uneven, or the minor contributor is submerged beneath major-contributor signal[5]. The National Institute of Standards and Technology (NIST) scientific foundation review states that uncertainty in contributor-number estimation increases as the number of contributors rises, allele overlap grows, and template amount falls, and further notes that low amounts of DNA generate stochastic variation and artifacts that complicate mixture interpretation[5]. In other words, even a well-validated PG system cannot recover information that has been biochemically blurred beyond what the data support. SCAnDi’s central premise is that physical separation can reduce the inferential dimensionality before computation begins: Instead of reconstructing unknown contributors from a pooled signal, one can work from clusters of single-source-like cell profiles or small-cell-pool consensus profiles[2,3,6,7].
However, this does not eliminate the need for statistical formalization. In the single-cell investigative genetics framework, the “credible genotype set” is the ranked set of locus genotypes whose posterior probabilities sum to at least 1-α; If genotypes are ordered by decreasing posterior probability, the credible set C1-α is the smallest set satisfying
Grgicak et al[7] used α = 0.002. Their 99.8% credible set captured the true genotype at 99.3% of loci across 630 mixtures containing up to five donors. This definition should be explicit because a credible set is not a “best guess”; it is a probability-bounded set whose practical utility depends on calibration, clustering quality, and the posterior model used to generate it[7,8].
A second boundary concerns proposition hierarchy. International Society for Forensic Genetics guidance, the Forensic Science Regulator, and more recent logical-framework papers all emphasize that sub-source findings cannot simply be carried upward to activity-level conclusions[22-26]. The 2025 Scientific Working Group on DNA Analysis Methods (SWGDAM) position statement on formal activity-level reporting made the same point in more operational terms, arguing that questions about “how” or “when” DNA was deposited raise scientific, practical, and legal difficulties that presently make routine United States implementation unsuitable[27]. This separation is not academic. Courts continue to evaluate forensic DNA evidence primarily through STR typing, LRs, and validated interpretation frameworks. Recent appellate decisions on continuous PG evidence have centered on whether STRmix™ or TrueAllele® were sufficiently validated and generally accepted for the mixture context at issue. They have not treated DNA sub-source findings alone as answers to transfer or timing questions[28]. SCAnDi-like workflows will therefore succeed only if they strengthen “who contributed this DNA?” without implying that they have automatically answered “how did it get there?” or “when was it deposited?”[23-25,27,28].
WHAT SCANDI ADDS AND WHAT IT DOES NOT
SCAnDi adds three genuinely important features beyond conventional bulk analysis. First, it preserves cell-of-origin information rather than destroying it at extraction[3]. Second, it explicitly seeks to separate cellular DNA from background DNA. Third, it incorporates phenotype-linked cell classification, which may constrain propositions with cellular context rather than genotype alone. These are meaningful additions, but they should be described with precision.
Background DNA refers to pre-existing human DNA on a substrate before the event of interest. It may derive from prior use, handling, environmental accumulation, or persistent low-level shedding from known or unknown individuals[29-32]. It is therefore a substrate-level condition, not a mechanism. Secondary transfer refers to a transfer pathway in which DNA moves from the original shedder to the target via an intermediate person or object[31,32]. Background DNA may include material that originally arrived by primary, secondary, or higher-order transfer, but the terms are not interchangeable. This distinction matters because isolation of intact cells may reduce co-amplification of substrate-level background material. It does not, however, establish the transfer route responsible for any recovered DNA[3,29,31,32].
The value of this distinction is supported by prevalence studies. Human DNA has been shown to be widespread on urban stones, household flooring, and other commonly touched surfaces, often producing mixed or difficult-to-attribute profiles[29-31]. SCAnDi’s attempt to separate intact cells from acellular or highly degraded substrate DNA is therefore biochemically plausible and forensically useful. Yet a strict “cell-only” strategy could also exclude probative extracellular DNA, which is relevant because touch and handled-item traces often contain both cellular and cell-free components[31,33]. Accordingly, SCAnDi-like workflows should begin with an explicit decision point: Whether the sample is being processed as a cellular-resolved evidence source, an acellular source, or a dual-fraction source with separate interpretive boundaries[3,31,33].
Phenotype-genotype linkage is potentially valuable, especially in sexual assault evidence where sperm and epithelial cells are intermingled and where body-fluid context affects proposition choice[3,34,35]. However, phenotype labels should not be treated as self-authenticating truth. Any imaging or classifier step that labels cells as sperm, vaginal epithelial cells, blood cells, or another cell type must itself be validated. Validation should include sensitivity, specificity, and misclassification rates under casework-like conditions, including aging, staining variation, substrate fluorescence, partial lysis, and mixed populations[3,6,34]. Without those data, cell typing risks becoming a new source of unquantified overconfidence rather than a constraint on interpretation[6,34].
WHAT THE STRONGEST SINGLE-CELL FORENSIC STUDIES ACTUALLY SHOW
The strongest recent validation of image-enabled digital cell sorting for forensics remains the 2024 DEPArray study by Schulte et al[6], which examined specificity, sensitivity, repeatability, and contamination for blood, epithelial, and sperm cells.
Diploid single-cell analysis yielded mean profile completeness of approximately 80% and mean peak heights around 290 relative fluorescence units (RFUs). Haploid sperm yielded approximately 51% completeness and mean peak heights around 176 RFUs. High stutter was a main, but recognizable, artifact.
The key operational finding was not merely that individual cells can be typed. Rather, complete or near-complete donor profiles often required consensus from more than one cell. In some settings, as few as three diploid cells recovered full profiles, whereas sperm required larger pools. Approximately 15 sperm cells were needed to exceed 90% completeness of the inferred autosomal diploid genotype. This is why “single-cell” should not be equated with routine one-cell completeness in casework reporting[6,9].
Furthermore, sexual assault applications provide a realistic stress test. Williamson et al[35] had already shown that DEPArray processing could improve purification of sperm and epithelial fractions compared with standard differential extraction in sexual offense samples. Schulte et al[34] extended this literature in 2025 by studying post-coital samples from 10 couples up to 96 hours after intercourse. They compared differential extraction, laser capture microdissection, and DEPArray-based workflows. That study found that time since intercourse and inter-individual variation strongly shaped male-cell recovery, and it also emphasized a practical limitation of cartridge-based imaging systems: Only a subset of loaded cells is scanned, commonly about 3000-6000 cells. Rare sperm can therefore be under-sampled at later time points or in female-dominated mixtures[34,35]. Crucially, this is not a minor engineering detail; it means that failure to detect male cells in the interrogated subset cannot be equated with absence of male DNA in the evidentiary substrate unless sampling probability has been quantified[34].
A simple approximation makes this operational point concrete: If minor-contributor cells constitute proportion (p) of the scanned cellular population and (n) cells are interrogated approximately at random, the probability of capturing at least one minor-contributor cell is 1 - (1 - p)n. Thus, if p = 0.001 and n = 3000, the probability is approximately 0.95. If only n = 1000 cells are scanned, it drops to approximately 0.63. Real casework will deviate from ideal random sampling because intactness, staining, gating, and morphology affect recoverability, but this minimal calculation shows why scan capacity and enrichment steps must be treated as part of evidential sensitivity rather than as invisible laboratory preliminaries[34].
Moreover, micromanipulation plus whole-genome amplification (WGA) occupies a different place on the workflow spectrum. Theunissen et al[36] showed that single sperm cells manually recovered from intimate swabs could, after amplification, yield more than 80% of haploid autosomal STR alleles in most individual sperm, and that Y-STR and X-STR data from single sperm cells could help sort autosomal haploid profiles into donor-specific diploid consensus genotypes in two-donor sperm mixtures. This study remains important because it demonstrates that physically isolating the contributor before inference can rescue donor information even when sperm are few, but it also underscores that operational usefulness frequently depends on combining autosomal STRs with lineage markers and consensus logic rather than relying on a single isolated electropherogram[9,36,37].
The most statistically mature downstream inference study to date is Grgicak et al[7], whose single-cell investigative genetics framework analyzed 630 mixtures containing up to five donors using direct-to-PCR single-cell electropherograms, suspect-agnostic clustering, and posterior genotype inference for database searching. The study reported that 99.3% of true genotypes were included in a 99.8% credible set. The most probable genotype was correct for 97% of loci when clusters contained at least two cells. Of 55418 locus-level posteriors, 86% returned only one credible genotype, and 99% of those singleton sets were correct. These are strong data, but they depend on three conditions that must remain visible in any translational claim: The donor distribution was constructed and calibrated, the smallest contributor contributed at least two cells, and the workflow assumed that clustered single-cell electropherograms could reasonably be treated as technical replicates from a single donor. That is promising forensic science, but it is not yet equivalent to universal casework readiness[7-9].
BIOCHEMICAL AND TECHNICAL BOTTLENECKS THAT WILL GOVERN FORENSIC VALUE
Single-cell forensic typing is governed by three biochemical constraints. First, template amount is minute: A diploid human cell contains approximately 6.6 pg of DNA and a sperm cell is haploid[2,6,36,37]. Second, early amplification events disproportionately shape artifacts, so severe imbalance, stutter, and dropout are expected rather than exceptional at low-template[5,6]. Third, any preamplification step, especially WGA, introduces method-specific representational bias across loci[36,37]. These realities mean that performance should be described locus-by-locus and cell-type-by-cell-type, not only as a global success percentage[6,36,37].
Allele designation deserves explicit treatment because it is a practical weak point in low-template cell-resolved analysis. NIST notes that peak positions are used to designate alleles, while peak heights become increasingly variable as template decreases; low-template amplification increases severe heterozygote imbalance, allele dropout, high stutter, and allele drop-in[5]. In single-cell workflows, this requires conservative locus calling rules. Put simply, low, isolated peaks that are not preserved across validated replicate or consensus logic should generate no-calls rather than forced allele designations[5,6,8]. Laboratories should publish the analytical threshold, the stochastic threshold if they use one, the replicate rule, the consensus rule, and the treatment of elevated stutter in cell-resolved data before any casework use[5,6,8,10].
WGA remains the most obvious leverage point for improving forensic utility. It is also a major source of bias. Classical forensic studies already showed that WGA can materially increase low-copy-number profiling success while simultaneously generating extra stutter and amplification bias[36,37]. In broader single-cell genomics, methods such as primary template-directed amplification were developed to improve uniformity, and benchmarking has continued to show systematic differences among single-cell amplification strategies[38-40]. These advances cannot simply be imported into forensic STR analysis by analogy. Forensic panels interrogate short repetitive loci, not only genome-wide single-nucleotide variation. Therefore, the best WGA method for whole-genome coverage is not necessarily the best method for balanced STR allele recovery[36-41]. Accordingly, any SCAnDi-like platform using WGA should report locus-specific dropout, imbalance, and stutter distributions on the exact forensic kit used in the intended workflow[6,36,37,41].
Sampling bias is the second bottleneck and is not solved by better chemistry alone. Methods that visually or algorithmically select cells can favor intact, stain-positive, morphologically obvious, or electrically responsive cells while excluding lysed, damaged, or extracellular material[6,31,34]. If the recovered subset departs systematically from the deposited population, then the cellular mixture ultimately typed may not reflect the biological mixture originally present on the substrate[31,34]. This is especially important for touch and contact traces, where donor shedding, substrate retention, prior background accumulation, and transfer history all influence what cells remain available for isolation[31-33]. The immediate implication is that negative single-cell findings should almost never be phrased as strong absence statements unless capture probability, enrichment efficiency, and acellular fractions have also been characterized[31,34].
Finally, contamination control becomes critical at near-single-template sensitivity. SWGDAM contamination guidance has long noted that human DNA is pervasive and that increasing assay sensitivity reveals contamination events that previously went undetected[42]. Single-cell pipelines add more opportunities for such events through staining, transfer, imaging, low-volume reaction setup, increased cycle sensitivity, replicate amplification, and post-PCR handling[6,42]. The appropriate standard is therefore not merely that “the negative control was clean”. It is whether the laboratory has quantified unexpected-allele and drop-in event rates for each workflow stage, documented upper confidence bounds for those rates, and pre-specified action limits for repeating or halting a batch when negatives show interpretable human signal or recurrent exogenous donor patterns[6,10,42].
IMPLEMENTATION, DATABASE UPLOAD, SCALABILITY, AND REPORTING BOUNDARIES
Casework entry should be governed by the same principles that apply to other forensic DNA methods, but even more strictly because the upstream analytical system is materially altered. SWGDAM validation guidance and the FBI Quality Assurance Standards state that validation shall precede implementation of any method used for forensic DNA analysis. Internal validation studies should include sensitivity and stochastic studies, mixture studies, contamination assessment, and casework-relevant samples[10,43]. A SCAnDi-like workflow is therefore a new forensic methodology even when it ends in a familiar capillary electrophoresis STR kit, because cell isolation, staining, image classification, direct-to-PCR processing, WGA, and cell-pool consensus modify both the data-generating process and the interpretation problem[6,10,37,43,44].
Database compatibility should also be stated with greater precision than is usually seen in proof-of-principle papers. Under current National DNA Index System (NDIS) procedures, a single-source forensic unknown or a fully deduced profile originating from a mixture is not intrinsically disqualified merely because it arose from mixture resolution; forensic DNA records generated through validated PG may also be submitted[11]. However, United States upload eligibility remains conditional. Forensic mixture and forensic partial records must have at least eight original Combined DNA Index System core loci and satisfy a moderate-stringency match-rarity threshold of 1 in 10 million. Single-source or fully deduced profiles are subject to allele-count constraints. Importantly, forensic records developed using low-template or low-copy-number analysis generally may not be submitted to NDIS outside missing-person or unidentified-remains contexts. Thus, a consensus profile from a single-cell or small-cell-pool workflow may be database-eligible only if the jurisdiction accepts the workflow as a validated casework method and the resulting profile satisfies the applicable interpretability and policy criteria[11,45]. This point should be made explicitly because “searchable” is a jurisdictional and quality-controlled term, not a synonym for “scientifically interesting”.
Against this backdrop, the implications for partial profiles are clear. If stochastic effects create locus-specific dropout, elevated stutter, or uncertain calls, the downstream product may still be probative for sub-source evaluation but may fail local database admission rules[5,6,11]. Accordingly, laboratories should validate two separate endpoints. The evidential endpoint asks whether the profile supports an LR-based evaluative or investigative inference under validated conditions. The database endpoint asks whether the profile meets national upload rules[11,12]. Collapsing these endpoints risks either overstating the casework value of partial cell-resolved profiles or understating their utility in instances where non-uploadable evidence still materially constrains contributor inference[5,11,12].
Scalability, cost, and workflow realism also deserve critical attention. Cartridge-based or image-enabled sorting systems remain lower-throughput than bulk extraction. This is especially true when only a few thousand cells can be scanned per run, manual image review is required, or replicate typing is needed to build consensus[6,34]. Consumable costs, clean-laboratory segregation requirements, staff training, software validation, and evidence-consumption pressures all increase further if multiple small pools or repeat analyses are required[6,10,43]. From an accreditation perspective, these are not peripheral inconveniences. They alter standard operating procedures, competency assessment, contamination surveillance, technical review burdens, and report wording. These changes occur within a quality system already strained by complex DNA interpretation and human-factors pressures[21,43,44]. For precisely these reasons, broad replacement of bulk STR casework is neither necessary nor realistic at present. The evidence instead supports deployment as a triage or salvage method for narrowly defined, high-value scenarios: Non-searchable high-order mixtures, selected sexual assault samples in which sperm are present but bulk fractions remain female-dominated, and rare cold cases in which additional cell-resolved information could justify the extra labor and evidence consumption[3,6,7,34].
Consequently, formal LR reporting should be pre-defined before the first contested case. SWGDAM’s 2025 LR-reporting guidance recommends that if a laboratory uses an upper LR cap, it should not be lower than one trillion, and it warns that any cap must not be mistaken for an identity threshold[12]. This is directly relevant to single-cell workflows because a highly probative cell cluster can generate an impressive LR without converting the evidence into source attribution certainty. In our view, SCAnDi-like reports should always state the proposition level, report the numerical LR where a comparison is performed, specify whether the result derives from a single cell, replicated single cells, or a defined small-cell pool, and explicitly caution that cell-type linkage does not itself answer activity-level questions[12,23,24,27].
The translational workflow and reporting boundary are summarized schematically in Figure 1.
Figure 1 Proposed one-panel schematic for the translational logic of single-cell and single-molecule analysis for deoxyribonucleic acid identification-like workflows.
Bulk short tandem repeat (STR)/probabilistic genotyping should be retained when it already answers the forensic question. In non-interpretable or high-value cases, a cell-resolved workflow may proceed through cell recovery and imaging, validated cell-type classification, single-cell or small-cell-pool STR typing, and clustering/consensus-based probabilistic interpretation, with database-compatible sub-source reporting where local criteria are met. A hard reporting boundary remains: Cellular resolution alone does not resolve transfer, persistence, prevalence, recovery, or timing, and activity-level evaluation requires separate transfer, persistence, prevalence, and recovery data and separate propositions. TPPR: Transfer, persistence, prevalence, and recovery; PG: Probabilistic genotyping; STR: Short tandem repeat.
The proposed validation and implementation benchmarks are summarized in Table 1. The thresholds in Table 1 are intentionally framed as implementation benchmarks rather than universal scientific constants; they translate the strongest current validation studies and quality-system requirements into a checklist that laboratories, regulators, and courts can interrogate before casework use[6,10-12,43].
Table 1 Proposed validation and implementation checklist for single-cell and single-molecule analysis for deoxyribonucleic acid identification-like forensic workflows.
Validation domain
Proposed minimum deliverable or benchmark for intended use
Cell recovery and sampling
Report the probability of recovering at least one target minor-contributor cell under casework-like conditions; if the workflow is used to support absence-sensitive reasoning, the validated capture probability for the claimed scenario should be explicitly stated
Cell-type classification
Publish sensitivity, specificity, positive predictive value, and misclassification rates by substrate, staining condition, and sample age; do not use phenotype labels to support source-level wording without validated error rates
Minimum pooling and consensus
Validate the minimum number of cells needed to reach the laboratory’s pre-specified completeness target; current evidence supports at least small diploid pools, and approximately 15 sperm cells to exceed 90% inferred autosomal diploid completeness in one validated DEPArray setting
Dropout and stutter
Publish locus-specific dropout and stutter distributions by cell type and workflow; define no-call rules for loci that fail consensus or replicate criteria
Contamination and drop-in
Quantify stage-specific unexpected-allele and drop-in rates; require no interpretable human profile in reagent blanks for routine release and pre-define batch action limits for recurrent exogenous signal
Reproducibility and robustness
Demonstrate reproducibility across operators, instruments, days, substrates, and biologically realistic mixtures; include degraded, aged, and background-rich samples rather than only clean artificial mixtures
Concordance with reference methods
Demonstrate concordance of recovered contributor genotypes against known reference samples and against bulk STR where comparison is informative
Database compatibility
Validate upload eligibility separately from evidential usefulness; document whether resulting profiles meet local locus-count, allele-count, and statistical requirements for database searching
Reporting boundaries
Pre-define permissible outputs at sub-source and limited source level; state that cellular resolution does not by itself resolve transfer, persistence, prevalence, recovery, or timing
The recommended reporting boundary for SCAnDi-like casework is summarized in the following paragraph.
Recommended reporting boundary for any SCAnDi-like casework output. Improved cellular resolution may strengthen sub-source inference and, in selected settings, limited source-level inference. It does not by itself resolve transfer, persistence, prevalence, or timing. Activity-level evaluation requires separate transfer, persistence, prevalence, and recovery data and separate propositions[24,25,27].
CONCLUSION
Single-cell forensic DNA typing has crossed the central feasibility threshold. However, the present literature remains weighted toward proof-of-principle studies, selected sample types, specialized laboratories, and workflows that often depend on replication, consensus, or small-cell pools. Routine complete one-cell genotypes are not yet the norm. The decisive translational question is therefore narrower and more practical than early enthusiasm sometimes suggested: Can a laboratory validate an end-to-end workflow that quantifies cell recovery, phenotype misclassification, locus-specific stochastic error, contamination, and database compatibility well enough to improve forensic reliability in narrowly defined scenarios? The best current answer is that such workflows are promising adjunctive tools for high-order mixtures, selected sexual assault evidence, and certain cold cases. They are not yet broad replacements for bulk STR analysis combined with established PG.
The immediate priority is not another isolated demonstration that a single cell can yield an STR profile. It is the publication and interlaboratory testing of explicit implementation standards, including validated minimum cell counts for consensus, stage-specific contamination rates, sample-type-specific capture probabilities, database-eligibility decision rules, and report language that prevents sub-source findings from drifting into unsupported activity-level claims. If those standards are met, SCAnDi-like technology will deserve a defined place in forensic genetics. If they are not met, increased cellular resolution will merely relocate uncertainty upstream rather than resolving it.
ACKNOWLEDGEMENTS
The authors thank forensic practitioners and academic investigators whose validation-focused work has made critical appraisal of single-cell pipelines possible.
UK Research and Innovation.
SCAnDi: Single-cell and single-molecule analysis for DNA identification. Gateway to Research, Project reference ES/Y010655/1.
[PubMed] [DOI]
Butler JM, Iyer H, Press R, Taylor MK, Vallone PM, Willis S.
DNA mixture interpretation. National Institute of Standards and Technology. U.S. Department of Commerce, 2024.
[PubMed] [DOI] [Full Text]
Gill P, Hicks T, Butler JM, Connolly E, Gusmão L, Kokshoorn B, Morling N, van Oorschot RAH, Parson W, Prinz M, Schneider PM, Sijen T, Taylor D. DNA commission of the International society for forensic genetics: Assessing the value of forensic biological evidence - Guidelines highlighting the importance of propositions: Part I: evaluation of DNA profiling comparisons given (sub-) source propositions.Forensic Sci Int Genet. 2018;36:189-202.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 61][Cited by in RCA: 87][Article Influence: 10.9][Reference Citation Analysis (0)]
Gill P, Hicks T, Butler JM, Connolly E, Gusmão L, Kokshoorn B, Morling N, van Oorschot RAH, Parson W, Prinz M, Schneider PM, Sijen T, Taylor D. DNA commission of the International society for forensic genetics: Assessing the value of forensic biological evidence - Guidelines highlighting the importance of propositions. Part II: Evaluation of biological traces considering activity level propositions.Forensic Sci Int Genet. 2020;44:102186.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 36][Cited by in RCA: 82][Article Influence: 13.7][Reference Citation Analysis (0)]
Kumari P, Chhikara A, Gupta V, Dalal J. Touch DNA in forensic science: a comprehensive overview and current status of the research.Egypt J Forensic Sci. 2026;16:23.
[PubMed] [DOI] [Full Text]
Specialty type: Biochemistry and molecular biology
Country of origin: Greece
Peer-review report’s classification
Scientific quality: Grade A, Grade B, Grade B, Grade C
Novelty: Grade B, Grade B, Grade B, Grade C
Creativity or innovation: Grade B, Grade B, Grade B, Grade C
Scientific significance: Grade B, Grade B, Grade B, Grade C
P-Reviewer: Cardona F, PhD, Assistant Professor, Senior Postdoctoral Fellow, Spain; Lisman D, Affiliate Associate Professor, Poland; Smith JH, PhD, Senior Researcher, South Africa S-Editor: Liu JH L-Editor: A P-Editor: Lei YY