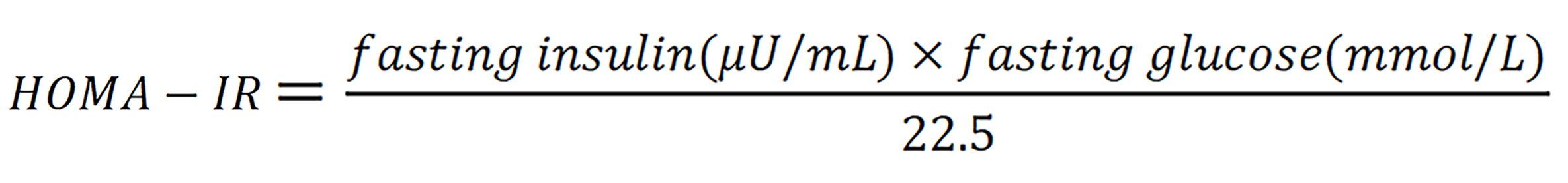Luo C, Zhang WW, Hua LY, Zeng MQ, Xu H, Duan CZ, Xu SY, Zhan S, Pan XF, Sun D, Ye LY, He DJ. Androgen receptor mutations in familial androgen insensitivity syndrome: A metabolic reprogramming pathway to type 2 diabetes susceptibility. World J Diabetes 2025; 16(11): 112236 [PMID: 41278448 DOI: 10.4239/wjd.v16.i11.112236]
Corresponding Author of This Article
Dong-Juan He, MD, Chief Physician, Dean, Director, Professor, Department of Endocrinology, The Second People’s Hospital of Quzhou, No. 338 Xin’an Avenue, Qujiang District, Quzhou 324002, Zhejiang Province, China. hedongjuan1247@wmu.edu.cn
Research Domain of This Article
Endocrinology & Metabolism
Article-Type of This Article
review-article
Open-Access Policy of This Article
This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Cheng Luo, Liang-Yan Hua, Cheng-Zheng Duan, Shi-Yu Xu, Department of Endocrinology, The Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou People’s Hospital, Quzhou 324000, Zhejiang Province, China
Wei-Wei Zhang, Department of Science and Education, The Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou People’s Hospital, Quzhou 324000, Zhejiang Province, China
Mei-Qi Zeng, Department of Ophthalmology, The Quzhou Affiliated Hospital of Wenzhou Medical University, Quzhou People’s Hospital, Quzhou 324000, Zhejiang Province, China
Hui Xu, Department of Hospital Management, Quzhou Hospital of Traditional Chinese Medicine, Quzhou 324000, Zhejiang Province, China
Shuo Zhan, Department of Endocrinology, The Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, Henan Province, China
Xiao-Fei Pan, School of Clinical Medical, Hangzhou Medical College, Hangzhou 311300, Zhejiang Province, China
Da Sun, Institute of Life Sciences and Biomedical Collaborative Innovation Center of Zhejiang Province, Wenzhou University, Wenzhou 325000, Zhejiang Province, China
Li-Ya Ye, Department of Gynecology, The Second People’s Hospital of Quzhou, Quzhou 324002, Zhejiang Province, China
Dong-Juan He, Department of Endocrinology, The Second People’s Hospital of Quzhou, Quzhou 324002, Zhejiang Province, China
Co-corresponding authors: Li-Ya Ye and Dong-Juan He.
Author contributions: Luo C and Zhang WW were responsible for the conceptualization, writing of the original draft, literature review and summary, they contributed equally to this article, they are the co-first authors of this manuscript; Hua LY and Zeng MQ undertook formal analysis and data validation; Xu SY and Duan CZ were in charge of methodology and investigation; Xu H and Zhan S handled supervision and project administration; Pan XF and Sun D took on literature review and figure preparation; Ye LY and He DJ played important and integral roles in the study design and manuscript preparation, including supervising the study design, providing critical revisions, and ensuring the integrity of the research findings; they contributed equally to this article, they are the co-corresponding authors of this manuscript; and all authors thoroughly reviewed and endorsed the final manuscript.
Supported by the Quzhou Science and Technology Plan Project, No. 2022K69.
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Corresponding author: Dong-Juan He, MD, Chief Physician, Dean, Director, Professor, Department of Endocrinology, The Second People’s Hospital of Quzhou, No. 338 Xin’an Avenue, Qujiang District, Quzhou 324002, Zhejiang Province, China. hedongjuan1247@wmu.edu.cn
Received: July 22, 2025 Revised: August 6, 2025 Accepted: September 22, 2025 Published online: November 15, 2025 Processing time: 116 Days and 0.2 Hours
Abstract
Familial androgen insensitivity syndrome (AIS), resulting from inherited mutations in the androgen receptor (AR) gene, has traditionally been examined within the framework of disorders of sex development. However, growing evidence indicates that AR dysfunction also disrupts systemic metabolic homeostasis, predisposing affected individuals to insulin resistance and type 2 diabetes mellitus. This article synthesizes recent advances in genetics, transcriptomics, and physiology to elucidate how AR mutations drive tissue-specific metabolic reprogramming in key organs, including pancreatic β-cells, skeletal muscle, liver, and adipose tissue. Particular attention is given to a newly identified familial AR variant (c.2117A>G; p.Asn706Ser), which not only broadens the known mutational spectrum of AIS but also underscores the clinical importance of early metabolic risk screening in this population. We further examine how pubertal stage, hormone replacement therapy, and sex-specific signaling pathways interact to influence long-term metabolic outcomes. Lastly, we propose an integrative management framework that incorporates genetic diagnosis, endocrine surveillance, and personalized pharmacological strategies aimed at reducing the risk of type 2 diabetes mellitus and cardiometabolic complications in individuals with AIS. Distinct from previous AIS-centered reviews, this work integrates metabolic and endocrine perspectives into the traditional developmental paradigm, offering a more comprehensive understanding of disease risk and translational management.
Core Tip: Familial androgen insensitivity syndrome (AIS), caused by androgen receptor gene mutations, has long been viewed as a disorder of sex development. However, recent findings revealed that androgen receptor dysfunction also drives metabolic reprogramming in key tissues - pancreatic β-cells, skeletal muscle, liver, and adipose tissue - leading to insulin resistance and increased susceptibility to type 2 diabetes mellitus. This article highlights emerging evidence of glucagon-like peptide-1 signaling disruption, mitochondrial dysfunction, and inflammatory imbalance in AIS. We propose an integrated framework of genetic screening, endocrine surveillance, and individualized therapy to improve long-term metabolic outcomes and prevent cardiometabolic complications in AIS patients.
Citation: Luo C, Zhang WW, Hua LY, Zeng MQ, Xu H, Duan CZ, Xu SY, Zhan S, Pan XF, Sun D, Ye LY, He DJ. Androgen receptor mutations in familial androgen insensitivity syndrome: A metabolic reprogramming pathway to type 2 diabetes susceptibility. World J Diabetes 2025; 16(11): 112236
Androgen insensitivity syndrome (AIS) is an X-linked hereditary disorder caused by loss-of-function mutations in the androgen receptor (AR) gene[1]. Because AR-mediated androgen signaling is impaired, 46, XY individuals become insensitive to endogenous androgens during development, resulting in absent or incomplete male sexual differentiation and, consequently, variably feminized phenotypes[2].
In recent years, type 2 diabetes mellitus (T2DM) and metabolic syndrome have spread rapidly worldwide, posing serious public health challenges[3]. By 2021, an estimated 537 million adults were living with diabetes globally. Obesity-related insulin resistance is a major precipitating factor for both T2DM and metabolic syndrome and markedly increases the risk of cardiovascular complications[4,5]. These epidemiological data underscore the urgent need to address metabolic imbalance and its downstream consequences.
Notably, the roles of androgen and AR signaling in metabolic homeostasis have attracted considerable attention. In addition to its high expression in reproductive organs, AR is abundantly expressed in key metabolic tissues - skeletal muscle, liver, adipose tissue, and pancreas - where it plays a pivotal role in regulating glucose and lipid metabolism[6]. Loss of AR signaling provokes pronounced metabolic derangements[6]. Mechanistic studies have shown that skeletal muscle-specific AR deletion diminishes glycolytic capacity and accelerates the onset of T2DM in male mice[7], whereas AR deficiency in pancreatic β-cells reduces glucose-stimulated insulin secretion (GSIS), leading to hyperglycemia and impaired glucose tolerance[8]. Collectively, these findings highlight the critical contribution of androgen/AR signaling to the systemic energy balance.
Given the profound metabolic influence of AR, a key question is whether AIS patients - i.e., individuals with congenital AR dysfunction - also face an elevated risk of metabolic abnormalities or diabetes. Previous research on AIS has focused mainly on sexual development and reproduction, but recent work has begun to examine its metabolic phenotype. Emerging reports suggest that AR loss of function may confer unfavorable metabolic characteristics in AIS patients[9,10].
This article aims to systematically delineate the mechanisms by which AR mutations influence glucose and lipid metabolism, with a particular focus on their roles in insulin resistance, dyslipidemia, and susceptibility to metabolic syndrome. A deeper understanding of androgen/AR signaling in metabolic homeostasis may offer new theoretical insights and clinical strategies for early identification and tailored management of metabolic disorders in the AIS population. While previous reviews have predominantly centered on genotype-phenotype correlations involving sexual development, gonadal dysgenesis, and psychosocial aspects, they have largely overlooked the metabolic consequences of AR dysfunction - especially its link to T2DM risk. In contrast, this review integrates emerging evidence from clinical observations, animal models, and tissue-specific AR knockout studies to construct a comprehensive framework connecting AR genotype with metabolic phenotype. To our knowledge, it is the first review to systematically explore this intersection and to propose a “genetics–metabolism-comorbidity” perspective on AIS, highlighting a pathophysiological dimension that has been underrecognized in the existing literature.
AR GENE MUTATIONS: GENETIC AND CLINICAL FEATURES OF AIS
AR gene structure and mutation spectrum
The human AR gene, located at Xq11-12, contains eight exons and encodes a nuclear receptor of approximately 920 amino acids[11,12]. Its architecture is highly modular: Exon 1 forms the N-terminal transactivation domain (carrying activation function 1); exons 2-3 constitute the DNA-binding domain with two zinc fingers that recognize androgen response elements; portions of exons 3-4 together create the hinge region, which mediates nuclear localization; and exons 4-8 encode the ligand-binding domain (LBD), which binds testosterone or dihydrotestosterone and houses activation function 2 for coactivator recruitment (Figure 1)[11,13].
Figure 1 Genomic location of the androgen receptor gene on the X chromosome and schematic structure of the encoded protein.
The upper panel shows the short (p) and long (q) arms of the X chromosome, with the androgen receptor (AR) gene located at Xq11-12. The middle panel illustrates the 5'→3'orientation of the AR gene, which contains eight exons (colored boxes). Exon 1 encodes the N-terminal transactivation domain (NTD), whereas exons 2-3 and 4-8 encode the DNA-binding domain (including zinc finger motifs) and ligand-binding domain (LBD), respectively. The lower panel displays the major structural domains of the AR protein: The NTD, which contains two polymorphic polyglutamine glutamine tracts; the DNA-binding domain, comprising two zinc fingers and a hinge region; and the LBD located at the C-terminus. Black arrows indicate the distribution of mutation sites identified in this study, which are predominantly clustered in the NTD and LBD. AR: Androgen receptor; NTD: N-terminal transactivation domain; DBD: DNA-binding domain; LBD: Ligand-binding domain; Gln: Glutamine; ZFM: Zinc finger motif; Gln(n): Glutamine (n repeats).
Because each segment fulfills a distinct function, impairment of any region can diminish AR activity and precipitate the AIS[14]. To date, more than 600 pathogenic variants associated with AIS have been identified - including missense, nonsense, splice site, regulatory region mutation, and exon deletion variants - distributed virtually across the entire AR gene[15,16]. Missense variants are the most common, typically clustering in the LBD or DNA-binding domain and disrupting ligand binding or DNA recognition, respectively; nonsense mutations and large deletions often generate truncated receptors or abolish ligand binding, usually producing the complete AIS (CAIS) phenotype. Variants in regulatory regions or deep introns leave the coding sequence intact yet attenuate receptor function by reducing transcription or altering splicing[17-19]. This complex and dispersed mutation landscape underlies the pronounced genetic and clinical heterogeneity of AIS, underscoring the need for deeper insight into AR structure-function relationships.
Epidemiology and functional prediction of familial AR variants
AIS is a rare X linked disorder; the incidence of complete CAIS is estimated to be approximately 1 20000-100000 46, XY newborns[14]. Most cases follow a X linked recessive pattern, with transmission from heterozygous carrier mothers; approximately two thirds are familial, whereas the remainder arise de novo. A considerable proportion of the latter represent early embryonic mosaic mutations, which may account for milder phenotypes or intrafamilial variability[20].
The mutation spectrum is highly diverse and largely consists of family specific private variants. Public databases - including androgen receptor gene mutations database, clinical variant database, and human gene mutation database - now catalog more than 800 pathogenic AR alterations, without evidence of a common founder mutation, making phenotype prediction challenging[14,21,22]. In general, missense variants within the LBD that retain partial ligand affinity tend to present as partial AIS (PAIS), whereas early truncating mutations or whole gene deletions more often cause CAIS; nevertheless, numerous exceptions exist[21].
In clinical practice, the pathogenicity of newly identified genetic variants is often evaluated via multiple in silico prediction tools - such as polymorphism phenotyping v2, sorting intolerant from tolerant, combined annotation–dependent depletion, and mutation tester - which assess sequence alterations on the basis of evolutionary conservation, physicochemical properties, and domain-specific localization[23]. These outputs are interpreted in accordance with the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, which classify variants into categories such as “likely pathogenic” or “benign”[24]. Although in vitro functional assays remain the gold standard, the integration of curated databases and predictive algorithms has become an indispensable aid in contemporary variant interpretation.
Clinical heterogeneity of AIS: Reproductive phenotypes, endocrine profiles, and familial aggregation
AIS is renowned for its marked clinical heterogeneity: Individuals with the same 46, XY karyotype can display a broad spectrum of phenotypes ranging from fully female to only mildly under masculinization[21,25]. AISs are traditionally classified into three forms.
CAIS: Phenotypically female with unambiguous female external genitalia. At puberty, breast development is normal, but menses are absent; pubic and axillary hair is sparse or lacking. Testes are present - typically intraabdominal or inguinal - and Mullerian structures are absent owing to anti-Mullerian hormone action. Laboratory findings usually reveal elevated testosterone and luteinizing hormone levels, reflecting targetorgan resistance. Virtually all CAIS patients harbor loss of function AR mutations[26,27].
PAIS: External genitalia are ambiguous or show intersex characteristics; partial masculinization occurs, with a small phallus, hypospadias, and often cryptorchidism. Pubertal development may tend toward either females or males, necessitating individualized sex assignment and surgical or hormonal management. Most PAIS associated mutations are missense variants in the LBD that preserve partial receptor function; however, the phenotype is modulated by multiple factors, leading to notable genotype-phenotype discordance[11,28].
Mild AIS: External genitalia are fully male, but after puberty, patients may exhibit mild masculinization, gynecomastia, or infertility (oligo/azoospermia). Mild AIS is often identified during infertility workups or family screening. Testosterone action is sufficient for male genital development, but receptor sensitivity is partially reduced[29].
Notably, members of the same family carrying an identical AR mutation can manifest CAIS, PAIS, or mild AIS, indicating that factors beyond the variant itself - such as fetal hormone exposure, AR coregulators, and epigenetic modifications - also influence phenotypic expression[26,30].
Preliminary evidence linking AR mutations to metabolic phenotypes
Recent studies have revealed that AR mutations not only affect sexual development but may also interfere with fat distribution, insulin sensitivity, and the risk of T2DM. Animal model studies have demonstrated that global AR knockout mice exhibit typical features of metabolic syndrome, including visceral obesity, insulin resistance, hepatic steatosis, and dyslipidemia. Tissue-specific AR knockouts have further uncovered its regulatory roles in the hypothalamus, liver, adipose tissue, and cardiovascular system[31,32].
In humans with AIS, although metabolic data remain limited, emerging trends are becoming increasingly evident. Some CAIS patient cohorts show tendencies toward increased body fat percentage, reduced insulin sensitivity, and lipid abnormalities. Despite the small sample sizes and confounding effects from estrogen replacement therapy, these findings suggest potential metabolic risks[33,34].
In the broader population, variations in AR cytosine-adenine-guanine trinucleotide repeat length - serving as a marker of functional polymorphism - have been significantly associated with metabolic syndrome, insulin resistance, and dyslipidemia[35,36]. Moreover, patients with AR-related disorders such as Kennedy’s disease (spinal and bulbar muscular atrophy) frequently exhibit glucose metabolism abnormalities and fatty liver, further supporting the link between reduced AR activity and metabolic dysfunction[37,38].
METABOLIC ABNORMALITIES AND DIABETES SUSCEPTIBILITY IN AIS PATIENTS
Case reports and cohort studies
The earliest evidence linking AIS to metabolic disturbances came from individual case reports and small series. Preclinical models and studies of oligozoospermic men have already shown that the loss of AR signaling promotes adiposity and insulin resistance, and clinical observations continue to support this relationship. Dati et al[9] analyzed 18 CAIS women (46, XY) and reported a markedly greater prevalence of obesity than controls did (16.7% vs 3.6%); most participants also presented abnormal insulin resistance indices. These early findings suggest that loss of AR function may increase body fat accumulation and disrupt glucose-lipid homeostasis.
More recent cases strengthen this association. In 2018, a 22-year-old Chinese CAIS patient who had undergone childhood gonadectomy and received no pubertal hormone replacement developed central obesity, dyslipidemia, and early-onset T2DM; combined therapy with insulin, metformin, and estradiol improved glycemic and lipid control, but body weight decreased only modestly[10]. This case illustrates how AR can precipitate an early metabolic syndrome profile, underscoring the key role of AR signaling in metabolic regulation. In 2023, a Malaysian CAIS patient with inadequate post pubertal hormone therapy progressed to extreme obesity [body mass index (BMI) > 50 kg/m²], obstructive sleep apnea, hypertension, fatty liver, and diabetes, ultimately requiring bariatric surgery - further evidence that poor hormonal management can expose AIS adults to severe metabolic risk[39].
Within a Chinese Han pedigree, our own work identified an AR variant, c.2117A>G (p.Asn706Ser); several carriers exhibited early visceral obesity and insulin resistance, supporting a direct link between AR deficiency and abnormal glucose metabolism. A recently published N706S family case and systematic review expanded the phenotypic and genetic evidence for this mutation[40]. To date, only a few population-based studies have evaluated metabolic outcomes in AIS patients.
The European disorders of sex development (DSD)-LIFE cross-sectional cohort (n = 222) reported that adolescents with 46, XY DSD - including AIS - had a mean BMI of 24 kg/m², with obesity and metabolic syndrome prevalences of 8% each, rates comparable to those of the general population; however, low bone mineral density (BMD) was observed in 15% of participants[41]. Although AIS represents the most common 46, XY DSD subtype in Asia (about 40%-80%), regional studies have focused mainly on phenotype-genotype correlations, with few systematic reports on insulin resistance, lipid profiles, or longitudinal cardiovascular outcomes[30,42-44]. In addition to scattered case reports and small cross-sectional studies hinting at mild metabolic risk, large longitudinal cohorts assessing the long-term metabolic trajectory of AIS patients are still lacking[10,45].
Metabolic differences between adolescent and adult AIS patients
The metabolic phenotype of AIS is strongly modulated by developmental stage and sex steroid exposure. Adolescent CAIS individuals who retain their testes can aromatize androgens to estrogens, thereby supporting female secondary sexual development and conferring partial metabolic protection; these individuals typically exhibit normal body weight and gynoid fat distribution, albeit with reduced lean muscle mass[14,34]. In PAIS, androgen supplementation during puberty, when some AR activity is preserved, can improve insulin sensitivity[46].
In adulthood, metabolic outcomes depend on the timing of gonadectomy and adherence to hormone replacement therapy (HRT). Early castration without prompt estrogen replacement predisposes patients to insulin resistance and central obesity[10]. In contrast, gonadectomy performed after puberty, followed by well structured estrogen replacement, is associated with more favorable metabolic profiles[33]. Transdermal estradiol and high dose testosterone exert comparable metabolic effects in CAIS, offering no clear advantage for androgen therapy[46]. The management of PAIS must be individualized: Androgens may be metabolically beneficial, but their use should be weighed against the degree of residual AR function[47]. Overall, maintaining adequate sex steroid levels is fundamental to optimizing body composition and metabolic health[48].
Interestingly, although AIS represents a state of androgen signaling deficiency, some middle-aged patients exhibit metabolic abnormalities resembling those observed in hyperandrogenic conditions such as polycystic ovary syndrome (PCOS), suggesting that both excess and deficiency of AR signaling can disrupt metabolic homeostasis[33,49]. Mechanistically, AR deficiency reduces muscle mass and glucose uptake, key drivers of metabolic dysregulation in AIS patients. Hence, AIS offers a unique human model for studying the interplay between sex hormones and metabolism[7,50]. Strategically timed gonadectomy and long-term, carefully managed HRT may prevent or mitigate adult onset metabolic complications in AIS patients (Table 1).
Table 1 Overview of the hormonal milieu, metabolic phenotypes, and key management points across developmental stages in androgen insensitivity syndrome.
Stage /subtype
Hormonal exposure and treatment strategy
Dominant metabolic phenotype
Key risk/protective factors
Management priorities
Adolescence - CAIS (testes retained)
High circulating testosterone → aromatized to estradiol; no hormonereplacement therapy (HRT)
Generally, normal body weight and lipid profile; feminine fat distribution; low lean (muscle) mass
Endogenous estradiol confers metabolic protection
Monitor bone mass and body composition; determine optimal timing of gonadectomy
Adolescence - PAIS (reared male + androgen supplementation)
Residual AR activity plus exogenous testosterone
↑ Muscle mass; possible improvement in insulin sensitivity
Partial restoration of androgen action
Tailor testosterone dose individually; guard against excess weight gain
MECHANISMS OF METABOLIC REPROGRAMMING TRIGGERED BY DEFECTIVE AR SIGNALING
AR loss in pancreatic β cells impairs insulin secretion and weakens glucagon-like peptide-1 signaling
AR is expressed in pancreatic β cells, where it augments insulin release through extranuclear mechanisms. In mice, β-cell-specific AR knockout (βARKO) markedly attenuates GSIS and impairs glucose tolerance[8,51]. Mechanistically, dihydrotestosterone activated AR, which is localized predominantly outside the nucleus, elevates intracellular cyclic adenosine monophosphate (cAMP), activates protein kinase A (PKA), and amplifies the insulinotropic action of glucagon-like peptide-1 (GLP-1). In vitro, dihydrotestosterone (DHT) enhances GLP-1 induced cAMP generation and insulin release, a process dependent on the GLP-1 receptor and AR mediated recruitment of stimulatory G alpha subunit proteins[51]. Conversely, βARKO mice exhibit blunted GLP-1 signaling, reduced cAMP/PKA responsiveness, and compromised Ca2+ dependent insulin exocytosis[52]. Transcriptomic analyses revealed the downregulation of GLP-1/cAMP pathway genes and the upregulation of inflammatory and secretory genes in AR deficient islets. MIN6 and INS1 β cell models likewise demonstrated that AR knockdown weakens GLP-1 evoked insulin secretion, whereas AR activation potentiates this response via the PKA-Ca2+ cascade[53]. Clinical observations concur: Men with androgen deficiency have poorer glycemic control, and AR antagonists in human islets dampen the insulinotropic effect of DHT. Overall, AR loss diminishes insulin secretory capacity by suppressing the GLP-1 receptor-cAMP/PKA axis and Ca2+ dynamics[54,55]. Although AIS models and induced pluripotent stem cell derived β like cells have only recently emerged, they are expected to manifest similar deficits in GLP-1 signaling (Figure 2).
Figure 2 Androgen receptor enhances insulin secretion in pancreatic β-cells via nongenomic coactivation of the glucagon-1 receptor-cyclic adenosine monophosphate/protein kinase A signaling axis.
Upon binding to the glucagon (GLP)-1 receptor on the plasma membrane, GLP-1 stimulates adenylate cyclase through a stimulatory G alpha subunit-dependent mechanism, leading to the generation of cyclic adenosine monophosphate (cAMP), activation of protein kinase A (PKA), and an increase in cytosolic Ca2+, which together trigger insulin secretion. The dihydrotestosterone-activated androgen receptor, which is predominantly localized outside the nucleus, further enhances this pathway by recruiting additional stimulatory G alpha subunit proteins to amplify adenylate cyclase activity, thereby potentiating GLP-1-cAMP-PKA signaling and promoting insulin release. In β-cell-specific androgen receptor knockout mice, this amplification loop is disrupted, resulting in impaired GLP-1 signaling, a reduced cAMP/PKA response, abnormal Ca2+ dynamics, attenuated glucose-stimulated insulin secretion, and ultimately glucose intolerance. AR: Androgen receptor; DHT: Dihydrotestosterone; GLP-1: Glucagon-like peptide-1; GLP-1R: Glucagon-1 receptor; AC: Adenylyl cyclase; cAMP: Cyclic adenosine monophosphate; PKA: Protein kinase A; GSIS: Glucose-stimulated insulin secretion; βARKO: Β-cell-specific androgen receptor knockout; Gαs: Stimulatory G alpha subunit; AIS: Androgen insensitivity syndrome; iPSC: Induced pluripotent stem cell.
Rewiring of insulin pathways and glucose dysregulation in skeletal muscle and liver
In skeletal muscle, AR enhances insulin sensitivity and glucose metabolism. Mice with muscle specific AR deletion develop early insulin resistance and a T2DM like phenotype; AR deficiency reduces glucose uptake and glycolysis by approximately 30%, accelerating diabetic progression[7]. In vitro, DHT increases glycolytic activity and glucose transporter type 4 (GLUT4) expression/translocation, whereas AR inhibition reverses this effect[56]. At the molecular level, AR directly regulates metabolic master genes such as PGC-1α and nuclear respiratory factor 1; its loss suppresses mitochondrial oxidative phosphorylation[7,57]. Consequently, mice with muscle specific AR deletion muscle shows lipid droplet accumulation, decreased fatty acid oxidation (despite the upregulation of β oxidation genes), increased amino acid catabolism, and oxidative stress. Systemically, these mice display reduced insulin tolerance, attenuated protein kinase B/phosphatidylinositol 3-kinase signaling, and decreased GLUT4 translocation[56,58].
In the liver, AR likewise modulates insulin signaling and glucose handling. Under a high fat diet, global or liver specific AR deletion exacerbates insulin resistance and impairs glucose tolerance, which coincides with the suppression of the insulin receptor (IR)/IR substrate pathway[31]. However, female liver specific AR deletion mice are partially protected from high fat induced metabolic derangements, indicating sex specific differences[59]. In AR deficient livers, steatosis activates protein kinase C epsilon, which further impairs IR function and diminishes IR substrate 1/2 activity. Orchidectomy or systemic AR knockout similarly blunts insulin action in muscle and liver, elevates tissue triglycerides, and reduces glycogen stores[56,59]. AR can also activate the adenosine monophosphate-activated protein kinase pathway, thereby promoting glucose uptake and utilization[60].
In summary, AR loss skews muscle and liver metabolism toward lipid accumulation and inflammation while suppressing insulin dependent glucose clearing mechanisms - namely, GLUT4 translocation and protein kinase B/adenosine monophosphate-activated protein kinase signaling (Table 2).
Table 2 Organ-specific differences in androgen - receptor signaling.
Organ/tissue
Principal action of AR
Metabolic consequences when AR signaling is impaired
Skeletal muscle
Drives glucose utilization and myofiber growth
AR loss impairs glycolysis and diminishes insulin sensitivity
Liver
Promotes fattyacid oxidation and enhances insulin responsiveness
AR deficiency predisposes to hepatic steatosis and insulin resistance
Adipose tissue
Restrains visceralfat accumulation and regulates lipid turnover
Lack of adipocyte AR increases visceral obesity and insulin resistance
Pancreatic βcells
Potentiates glucosestimulated insulin secretion
AR activation boosts insulin release, whereas AR knockout blunts excessive secretion
Hypothalamic neurons
Preserves wholebody insulin sensitivity
Neuronal AR deletion elicits hypothalamusdriven insulin resistance
AR preserves mitochondrial function and dampens inflammation in adipose tissue
AR sustains mitochondrial homeostasis and suppresses chronic inflammation in adipose tissue[61]. In adipocyte specific AR knockout mice, fat pads are reduced under a standard diet, yet fasting hyperinsulinemia is evident; when fed a high fat diet, these animals develop visceral obesity, hyperglycemia, and β cell failure[62]. Both adipose tissue and plasma contain elevated levels of retinol binding protein 4 (RBP4), a key adipokine that promotes insulin resistance. In vitro, AR activation represses RBP4 expression, whereas AR deficiency increases RBP4 expression[57,62].
At the mitochondrial level, AR induces the expression of biogenic regulators such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and uncoupling protein 1 (UCP1). In brown/beige adipocytes, AR signaling inhibits thermogenesis by blocking cAMP response element-binding protein phosphorylation at the UCP1 promoter. Loss of AR lifts this repression, driving “browning” of white fat and increasing proton-leak respiration[63]. Conversely, AR deficiency disrupts lipid metabolism and reduces the mitochondrial content in white adipose tissue.
AR also exerts anti-inflammatory effects: DHT-activated AR attenuates nuclear factor kappa B (NFκB) signaling and suppresses proinflammatory cytokines such as tumor necrosis factor α and monocyte chemoattractant protein-1. In contrast, AR loss activates NFκB, leading to M1macrophage infiltration, chronic adipose inflammation, and systemic insulin resistance[57]. Correspondingly, AR null mice display elevated circulating interleukin-1β and upregulated NFκB target genes in adipose tissue[57,61]. In summary, AR maintains metabolic equilibrium in fat by modulating PGC1α/UCP1 dependent mitochondrial function and by curbing NFκB-mediated inflammation; its absence leads to lipid dysregulation, proinflammatory cytokine release, and diminished insulin sensitivity.
Gene epigenetic regulation and interplay with sex dimorphic effects
In addition to its acute signaling role, AR functions as a chromatin bound transcription factor, recruiting histone modifying enzymes and microRNA (miRNA)-regulatory complexes to mediate long range gene control. AR can bind directly to promoters of metabolic genes such as PGC1α, transcription factor a, mitochondrial, and phosphoenolpyruvate carboxykinase, modulating their histone acetylation and DNA-methylation status[64]. It likewise governs non-coding RNAs, including the miR-200/375 families, which participate in metabolic pathways in muscle, liver, and β-cells[65,66].
Conversely, sex-steroid pathways reshape the AR epigenetic landscape: Estrogen signaling alters AR methylation and chromatin conformation, generating sex-specific AR action profiles[67]. Both clinical and animal data show that metabolic defects caused by AR loss are more pronounced in males. Male muscle-specific androgen receptor knockout mice develop a metabolic syndrome phenotype, whereas females maintain normal glycemic control[7,56]. A similar pattern is observed in the liver: Female liver-specific androgen receptor knockout mice retain insulin sensitivity on a high-fat diet, whereas males rapidly become insulin resistant[31]. This disparity likely reflects crosstalk between the AR and estrogen receptor (ER) pathways - male muscle metabolism depends heavily on the AR-PGC1α axis, whereas females preferentially engage in ERα signaling[7,68].
At the metabolic level, AIS patients (46, XY karyotype with complete AR loss) phenotypically resemble hypogonadal males, displaying increased adiposity and insulin resistance - further highlighting the central role of AR in male metabolic homeostasis[33]. To dissect these mechanisms at the cellular level, induced pluripotent stem cell-derived β-like cells and adipocytes carrying AR mutations offer promising models for future investigation[69]. In parallel, single-cell transcriptomic studies have uncovered cell type specific AR regulatory profiles in key metabolic organs such as myofibers and hepatocytes in males[70]. To integrate these findings, we summarize in Table 3 the tissue-specific roles of AR in glucose and lipid metabolism, along with their implications for T2DM susceptibility[7,31,46,51,57,71].
Table 3 Tissue-specific metabolic alterations associated with androgen receptor dysfunction in androgen insensitivity syndrome and their relevance to type 2 diabetes mellitus susceptibility.
Target tissue/system
AR-mediated function (normal)
Effect of AR dysfunction in AIS
Associated metabolic consequences
Ref.
Pancreatic β-cells
Promotes β-cell mass, insulin transcription, and GLP-1 sensitivity
GLP-1 signaling in T2DM and sex-specific differences
GLP-1 is an incretin hormone that enhances GSIS and suppresses glucagon release, playing a central role in glucose homeostasis[72]. Impaired GLP-1 secretion and reduced receptor sensitivity have been observed in individuals with T2DM, contributing to β-cell dysfunction and postprandial hyperglycemia[73]. While GLP-1 signaling operates independently of the AR pathway, it may be indirectly affected in AIS due to altered gut hormone dynamics, adipokine profiles, or secondary estrogen exposure[74].
Sex differences in GLP-1 pharmacodynamics are increasingly recognized. Women have shown greater glycemic and weight responses to GLP-1 receptor agonists (GLP-1RAs) such as semaglutide and liraglutide, as well as a higher incidence of gastrointestinal side effects[75]. These effects are attributed to differential gastric emptying rates, GLP-1 receptor expression, and hormone interactions. In the context of AIS, where AR signaling is absent or impaired, these sex-based metabolic responses may be further modified. Thus, GLP-1RAs represent a theoretically attractive option for managing obesity and T2DM in AIS, particularly given their mechanism does not rely on AR functionality[76].
CLINICAL EVALUATION AND METABOLIC RISK SURVEILLANCE IN AIS PATIENTS
Genetic testing for AR mutation and pathogenicity grading
The molecular diagnosis of AIS depends chiefly on thorough interrogation of the AR gene. Single gene sequencing by Sanger or next generation sequencing can detect approximately 95%-97% of pathogenic AR variants[13]. To capture the remaining 3%-5%, multiplex ligation dependent probe amplification, quantitative polymerase chain reaction, or array-based platforms based platforms are required to identify multiexonic or whole gene copy number variants[2,77]. When routine testing is negative, whole exome sequencing or whole genome sequencing can be employed to uncover rare intronic or regulatory region alterations[18]. Recent studies indicate that approximately 18% of AIS diagnoses depend on whole exome sequencing/whole genome sequencing, with reports of deep intronic mutations in PAIS and 5’UTR variants in both CAIS and PAIS[19,78]. Accordingly, current guidelines recommend pairing exon sequencing with copy number variant analysis and, in challenging cases, extending the assay to promoters, splice sites, and other noncoding regions[79].
Use of glucose tolerance test, homeostatic model assessment of insulin resistance, and lipid omics in metabolic assessment
Although patients with AIS exhibit a distinctive hormonal profile, accumulating evidence indicates that they may nevertheless develop disorders of glucose and lipid metabolism[10]. Some CAIS individuals present with central obesity, insulin resistance, and dyslipidemia during adolescence or adulthood, and several case reports have described marked improvements in glycemic and lipid indices after combined treatment with insulin, metformin, and estrogen[39]. Similarly, cross sectional studies have shown that CAIS is frequently accompanied by increased adiposity, elevated total cholesterol and low-density lipoprotein (LDL), and abnormal fasting insulin resistance - as measured by fasting insulin resistance [homeostatic model assessment of insulin resistance (HOMA-IR)], where
These findings suggest that the loss of AR signaling conveys a metabolic risk profile reminiscent of androgen deprivation[33].
Accordingly, AIS individuals should undergo systematic metabolic surveillance beginning at puberty, including an OGTT, HOMA-IR assessment, and routine lipid panels, with annual follow up thereafter[10]. This recommendation is particularly pertinent for Asian populations - especially Chinese patients - who can develop insulin resistance at comparatively low BMI thresholds, warranting even earlier intervention[80]. Although no AIS specific lipidomic studies have yet been reported, mass spectrometry analyses in and Rogen related disorders such as PCOS have identified numerous aberrant lipid species, providing a promising direction for future AIS biomarker discovery[81,82]. Currently, however, conventional glucose and lipid testing remains an effective means of detecting metabolic abnormalities in the AIS population.
Recent studies suggest that AR signaling interacts with intracellular second messenger pathways, notably the cAMP-PKA axis. AR activation modulates the expression and activity of adenylyl cyclases and phosphodiesterases, thereby influencing cAMP turnover in insulin-sensitive tissues such as adipose and muscle. In AR-deficient models, reduced PKA activity has been linked to impaired lipolysis and altered thermogenic responses, particularly through dysregulation of PKA targets like hormone-sensitive lipase and UCP1[51].
Moreover, GLP-1 signaling - which relies on cAMP and PKA to enhance insulin secretion and promote β-cell survival - may be indirectly affected by AR dysfunction. While GLP-1 receptor activation itself is androgen-independent, crosstalk between AR signaling, sex steroids, and cAMP responsiveness is increasingly recognized. In pancreatic β-cells, testosterone acting via AR amplifies GLP-1-induced insulin secretion via the cAMP-PKA pathway; this effect is blunted in βARKO models, which show reduced incretin-induced insulin secretion and diminished PKA activation despite intact GLP-1 receptor expression[51,52,83]. These findings suggest that AR dysfunction may impair the metabolic efficacy of GLP-1, especially in settings such as AIS, where AR signaling is compromised.
Metabolic-monitoring protocol before and after HRT
Patients with AIS require lifelong endocrine follow-up, particularly during the initiation of HRT[84]. Most CAIS individuals (female phenotype) undergo gonadectomy after puberty and subsequently receive estrogen replacement; a subset of these individuals has also been included in comparative studies in which testosterone was used as a prohormone[46]. PAIS patients who are male generally receive androgen therapy during puberty[47].
A recent randomized crossover trial demonstrated that six months of either transdermal estradiol or testosterone in CAIS women increased BMI by about 2.7% and worsened lipid profiles (higher total cholesterol and LDL, lower high-density lipoprotein), indicating that HRT - regardless of hormone type - may aggravate metabolic risk[46]. Accordingly, body weight, waist circumference, blood pressure, fasting glucose, and serum lipids should be reassessed 3-6 months after HRT initiation and annually thereafter[85]. Insulin sensitivity (HOMA-IR or OGTT) should likewise be monitored, especially in obese or high-risk patients[86].
For pubertal induction, follow-up every 3-6 months is recommended initially to titrate the dosage; once a maintenance regimen is established, visits may be reduced to annual intervals. One tertiary center advises reviews every 2-6 months during the early phase, with yearly checks once HRT is stable[87]. The following parameters were investigated: Body composition [dual-energy X-ray absorptiometry (DXA) or bioelectrical impedance], blood pressure, fasting glucose/Lipid/Liver enzymes, and HOMA-IR. In individuals with obesity or dyslipidemia, hepatic evaluation by ultrasound or elastography every 2-3 years is recommended to screen for nonalcoholic fatty liver disease (NAFLD)[88]. Tailoring the surveillance schedule - more frequently for high-risk patients - facilitates early identification of metabolic complications.
The clinical management of AIS should extend beyond HRT to include comprehensive assessments of skeletal, metabolic, cardiovascular, and hepatic health[89]. Androgen deficiency may impair bone mineralization; thus, delayed or inadequate HRT can lead to reduced BMD and an elevated risk of fractures. It is recommended that a baseline DXA scan be performed in late adolescence or early adulthood. If bone loss is detected, follow-up DXA should be conducted every 2-3 years, alongside calcium and vitamin D supplementation and encouragement of weight-bearing exercise to support bone health[85].
Cardiovascular risk screening is equally crucial. AIS patients should undergo annual monitoring of blood pressure, fasting glucose, and lipid profiles. In cases of insulin resistance or dyslipidemia, stricter therapeutic targets should be implemented. For older patients or those with additional risk factors, noninvasive vascular assessments such as carotid intima-media thickness or pulse wave velocity may be considered[14].
NAFLD is also relatively common in AIS individuals with coexisting obesity or glucose dysregulation. Periodic hepatic ultrasound or elastography should be conducted to detect steatosis and fibrosis at an early stage[90]. Given that NAFLD is often associated with elevated BMI and insulin resistance, it should be incorporated into the routine metabolic surveillance of AIS patients.
Optimal AIS care requires multidisciplinary collaboration involving endocrinology, gynecology or urology, clinical genetics, metabolic medicine, and psychological support. This approach addresses challenges related to gender identity, somatic changes, and chronic disease burden[91,92]. Expert consensus recommends lifelong follow-up, with regular assessments of body composition and cardiovascular risk as patients age. Integrating BMD evaluation, metabolic and vascular monitoring, and hepatic imaging into a genotype-phenotype-treatment–based personalized care plan may significantly enhance long-term health outcomes for individuals with AIS (Table 4)[65,85].
Table 4 Metabolic surveillance parameters and assessment methods for androgen insensitivity syndrome patients.
Monitoring parameter
Assessment method(s)
Purpose/clinical significance
Glucose tolerance
Fasting plasma glucose
Detect impaired glucose tolerance and stratify risk of T2DM
OGTT
HbA1c
Insulin resistance
HOMAIR = (fasting insulin × fasting glucose)
Quantifies insulin sensitivity (clamp technique is the gold standard)
Hyperinsulinemic–euglycemic clamp
Lipid profile
Serum TG, LDL, HDL, etc.
Identifies dyslipidemia and gauges metabolicsyndrome risk
Body composition
BMI
Assesses degree of obesity and fat distribution
Waisttohip ratio
DXA for bodyfat percentage
Bone mineral density
DXA of axial skeleton
Monitors bone mass and helps prevent osteoporosis (particularly important after gonadectomy in AIS)
Lifestyle factors
Dietary pattern analysis
Complements metabolic risk assessment and guides targeted interventions
Emerging evidence suggests that dysregulation of androgen and estrogen signaling can indirectly alter calcium and vitamin D homeostasis, which are critical for mitochondrial function and skeletal health[93,94]. ERs α/β and AR have been identified in osteoblasts and renal tubules, where they modulate calcium absorption and 1α-hydroxylase expression - the enzyme responsible for converting inactive vitamin D into its active form calcitriol [1,25(OH)2D][95]. Hypoandrogenism in AIS may thus compromise vitamin D activation, leading to secondary calcium imbalance. Furthermore, vitamin D itself influences mitochondrial respiration and reactive oxygen species production in muscle and adipose tissues. Impaired vitamin D signaling may exacerbate mitochondrial dysfunction in AIS, particularly under AR or ERs deficiency[95,96]. In animal models, vitamin D deficiency was shown to reduce mitochondrial oxidative phosphorylation and biogenesis via suppression of PGC-1α and nuclear respiratory factor 1/2 pathways[96].
INTERVENTION STRATEGIES AND PERSONALIZED MANAGEMENT IN AIS
AIS is a lifelong condition requiring personalized, stage-specific management that extends beyond gonadal development. The natural history of AIS includes sex assignment in infancy or early childhood, gonadectomy timing (typically after puberty in CAIS), pubertal induction, and long-term hormone replacement. In recent years, emerging evidence has emphasized the importance of addressing metabolic, cardiovascular, and psychological comorbidities in parallel. This section outlines current hormone therapies and metabolic interventions, followed by a proposed integrated care model for AIS across the lifespan.
HRT and its impact on metabolic outcomes
HRTs in patients with AIS should be tailored according to the rearing sex and residual AR functionality. Individuals with CAIS typically undergo gonadectomy after puberty, followed by estrogen replacement (commonly oral or transdermal estradiol at 1.5-2 mg/day) to maintain female secondary sexual characteristics[87,97]. While most CAIS patients use estrogen, some also trial testosterone therapy to enhance libido and quality of life[98,99].
A recent randomized crossover study revealed comparable metabolic safety between estrogen and testosterone, with both treatments leading to an average about 2.7% increase in BMI, elevated total cholesterol and LDL, and reduced high-density lipoprotein, although no significant short-term changes in insulin sensitivity were observed[46,97]. These findings underscore the need for routine monitoring of body weight, glycemia, and lipid profiles following sex steroid therapy in AIS patients.
Compared with oral formulations, transdermal estrogen is preferred for long-term use because of its avoidance of hepatic first-pass metabolism and lower thrombotic risk. If testosterone is chosen, short-acting transdermal or intramuscular preparations are recommended to mimic physiological conditions, with target hormone levels adjusted according to the rearing sex (Table 5)[100,101].
Table 5 Framework for individualized management and intervention pathways in androgen insensitivity syndrome.
Management/intervention domain
Core measures
Notes/targets
Diagnosis and genetic counseling
AR gene sequencing
Confirm AIS subtype and hereditary risk; inform reproductive and parenting decisions
Karyotype analysis
Familybased genetic counseling
Gonadal management (tumor surveillance)
Periodic ultrasound/MRI followup
Lower the risk of gonadal malignancy while allowing testicular hormones to support skeletal maturation
Elective orchiectomy after puberty
HRT
Initiate estradiol after puberty or postorchidectomy in CAIS
Maintain secondary sexual characteristics and prevent complications such as osteoporosis
Androgen therapy in PAIS as indicated
Bonemetabolic and cardiometabolic care
Regular monitoring of bonemineral density, blood glucose, and lipid profile
Control body weight, enhance insulin sensitivity, and protect against osteoporosis
Personalized advice on diet and exercise
Psychological and social support
Multidisciplinary team assessment
Facilitate genderidentity adaptation and mitigate psychological distress
Professional psychological counseling
Scheduled followup
Tracking of growth parameters, endocrine hormones, and tumor markers
Adjust therapy dynamically to achieve truly individualized care
As a monogenic condition, AIS presents a theoretical target for curative therapies such as gene editing, gene replacement, or molecular chaperones[102]. Although no such therapies are currently approved for clinical use in AIS, proof-of-concept studies have shown that CRISPR/Cas9-mediated correction of AR mutations is feasible in cell models[103]. Additionally, small-molecule chaperones that enhance the folding or nuclear translocation of partially functional AR proteins are under investigation[104]. Limitations include delivery challenges, off-target effects, and the need for tissue-specific targeting, particularly for central nervous and reproductive tissues. While still experimental, these approaches may offer future therapeutic potential and justify ongoing basic research[105].
Evidence-based use of metformin, GLP-1RA, and sodium-glucose cotransporter 2 inhibitors
Although no large-scale clinical trials of antidiabetic agents have been conducted specifically in AIS patients, treatment decisions can draw upon data from populations with PCOS and functional hypogonadism. For AIS patients with obesity, insulin resistance, or glucose dysregulation, metformin is the first-line therapy for improving insulin sensitivity and glycemic control[106].
GLP-1RAs (e.g., liraglutide and semaglutide) have demonstrated superior efficacy in lowering glycemia and reducing weight in PCOS patients and may be considered for obese AIS patients[107,108]. Sodium-glucose cotransporter 2 (SGLT-2) inhibitors (e.g., dapagliflozin and empagliflozin), which promote urinary glucose excretion to control weight and blood glucose, also hold promise for AIS individuals with obesity or type 2 diabetes[109].
Since the metabolic effects of these agents are independent of the AR signaling pathway, their efficacy in AIS is unlikely to be compromised. However, potential adverse events - such as gastrointestinal discomfort (GLP-1RAs), urinary tract infections, and euglycemic ketoacidosis (SGLT-2 inhibitors) - should be carefully monitored during treatment.
Although AIS is a distinct genetic disorder, T2DM management principles may align with those used in individuals with functional hypogonadism or androgen deficiency[110]. Importantly, insulin resistance in AIS is often accompanied by altered adipose distribution and sex hormone imbalance, which can affect treatment response. Metformin remains the first-line agent due to its insulin-sensitizing effects and cardiovascular safety. In cases requiring additional therapy, GLP-1RAs and SGLT-2 inhibitors are effective for both glycemic control and weight loss, and their mechanisms - being independent of AR signaling - make them suitable for AIS[111-113]. However, HRT must be coordinated with antidiabetic treatment, as changes in estrogen or testosterone levels can affect lipid metabolism, insulin sensitivity, and body composition. Regular glycated hemoglobin monitoring, fasting lipid panels, and anthropometric assessments are essential[46,114,115]. Multidisciplinary coordination among endocrinologists, metabolic specialists, and dietitians is recommended to ensure optimal outcomes.
Stepwise management pathway for AIS patients with comorbid obesity
For individuals with a BMI ≥ 30 kg/m² or those with metabolic complications, a tiered intervention strategy should be implemented. The initial step involves the adoption of a structured dietary regimen - such as the Mediterranean diet or a low-glycemic-index diet - combined with a consistent exercise program consisting of at least 150 minutes per week of moderate-intensity aerobic and resistance training. Cognitive behavioral therapy should be incorporated to enhance treatment adherence and self-identity[116].
If lifestyle modifications prove insufficient, pharmacological weight-loss therapies may be considered on the basis of individual characteristics, including GLP-1RAs, orlistat, or other appetite-suppressing agents[117]. In cases of severe obesity or pronounced metabolic dysregulation, bariatric surgery (e.g., sleeve gastrectomy or gastric bypass) may be appropriate, contingent upon the preoperative evaluation of psychological readiness and postoperative HRT planning[118,119]. Postsurgical follow-up should include close monitoring of vitamin and hormone levels, with timely adjustments in estrogen formulations to ensure optimal bone metabolism and symptom management.
Integrated “DSD-metabolism-psychology-nutrition” follow-up model
Patients with AIS should receive life-course, multidisciplinary follow-up care. Endocrinologists oversee HRT, BMD monitoring via DXA, and annual metabolic screening (BMI, fasting glucose/glycated hemoglobin, lipid profile). Genetic counseling supports familial decision-making and variant interpretation. Dietitians provide individualized nutritional guidance to support pubertal growth and adult weight control. Metabolic specialists monitor insulin resistance and the risk of developing diabetes. Psychological support is essential throughout, addressing emotional well-being, gender identity, social adaptation, and concerns related to reproductive and sexual health[89,120,121].
Follow-up priorities should be tailored to developmental stages: In childhood, emphasis should be placed on growth and gender identity formation; during adolescence, the focus should shift to pubertal induction and HRT transition; and in adulthood, the effectiveness of HRT, cardiovascular risk, and psychological well-being should take precedence[99,115].
Efficient interdisciplinary communication is crucial - dietitians should be aware of how hormonal dosage changes affect body weight, whereas mental health professionals need to understand how physical changes may impact self-esteem. Establishing a shared health record system can facilitate the implementation of this integrative model[122].
CONCLUSION
AIS, a prototypical DSD in individuals with a 46, XY karyotype, is fundamentally driven by loss-of-function mutations in the AR gene. In recent years, accumulating evidence has revealed that AR signaling deficiency extends far beyond impaired sexual differentiation - it also disrupts systemic metabolic homeostasis, constituting a key genetic basis for insulin resistance, dyslipidemia, and even type 2 diabetes in AIS individuals. AR plays tissue-specific metabolic regulatory roles in pancreatic β-cells, skeletal muscle, the liver, and adipose tissue. Its dysfunction leads to metabolic pathway reprogramming and chronic low-grade inflammation, exacerbating glycemic dysregulation and lipid abnormalities.
In light of the increasing recognition of metabolic risk in AIS, there is an urgent need to establish an integrated precision assessment system that links genotype–clinical phenotype-metabolic indicators. This should include pathogenicity grading of AR mutations, dynamic monitoring of OGTT and HOMA-IR, lipid profiling, and body composition analysis, aiming to enable early identification of metabolic disturbances and guide individualized intervention strategies. In parallel, optimizing HRT, judicious incorporation of metabolic agents (e.g., metformin, GLP-1RAs, SGLT-2 inhibitors), and sustained multidisciplinary support are essential pillars for managing AIS-related metabolic abnormalities.
In the future, we advocate for the development of national or regional AIS-metabolism collaborative networks, the establishment of longitudinal follow-up cohorts and digital management platforms across the life course, and the exploration of emerging interventions such as AR function enhancement, gene editing technologies, and stem cell-based regenerative therapies. Tailored clinical guidelines should also be formulated to reflect the unique characteristics of local populations.
By bridging basic science with clinical translation, the construction of a scientifically grounded, standardized, and precision-oriented AIS metabolic health management framework will not only improve long-term outcomes and quality of life for affected individuals but also expand the scope of DSD care from gender identity to a more holistic model encompassing gender-metabolism-psychological well-being.
Zhang H, Zhou XD, Shapiro MD, Lip GYH, Tilg H, Valenti L, Somers VK, Byrne CD, Targher G, Yang W, Viveiros O, Opio CK, Mantzoros CS, Ryan JD, Kok KYY, Jumaev NA, Perera N, Robertson AG, Abu-Abeid A, Misra A, Wong YJ, Ruiz-Úcar E, Ospanov O, Kızılkaya MC, Luo F, Méndez-Sánchez N, Zuluaga M, Lonardo A, Al Momani H, Toro-Huamanchumo CJ, Adams L, Al-Busafi SA, Sharara AI, Chan WK, Abbas SI, Sookoian S, Treeprasertsuk S, Ocama P, Alswat K, Kong AP, Ataya K, Lim-Loo MC, Oviedo RJ, Szepietowski O, Fouad Y, Zhang H, Abdelbaki TN, Katsouras CS, Prasad A, Thaher O, Ali A, Molina GA, Sung KC, Chen QF, Lesmana CRA, Zheng MH. Global burden of metabolic diseases, 1990-2021.Metabolism. 2024;160:155999.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 282][Cited by in RCA: 242][Article Influence: 121.0][Reference Citation Analysis (0)]
Xu W, Qadir MMF, Nasteska D, Mota de Sa P, Gorvin CM, Blandino-Rosano M, Evans CR, Ho T, Potapenko E, Veluthakal R, Ashford FB, Bitsi S, Fan J, Bhondeley M, Song K, Sure VN, Sakamuri SSVP, Schiffer L, Beatty W, Wyatt R, Frigo DE, Liu X, Katakam PV, Arlt W, Buck J, Levin LR, Hu T, Kolls J, Burant CF, Tomas A, Merrins MJ, Thurmond DC, Bernal-Mizrachi E, Hodson DJ, Mauvais-Jarvis F. Architecture of androgen receptor pathways amplifying glucagon-like peptide-1 insulinotropic action in male pancreatic β cells.Cell Rep. 2023;42:112529.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 6][Cited by in RCA: 28][Article Influence: 9.3][Reference Citation Analysis (1)]
Helsen C, Rocca MS, Nguyen TT, Eerlings R, Lee XY, De Block S, Vinanzi C, Di Millo F, Giagulli V, Voet A, Ferlin A, Claessens F. Study of novel androgen receptor V770 variant in androgen insensitivity syndrome patients reveals the transitional state of the androgen receptor ligand binding domain homodimer.Protein Sci. 2023;32:e4599.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 2][Cited by in RCA: 6][Article Influence: 2.0][Reference Citation Analysis (1)]
Stella S, Vitale SR, Massimino M, Martorana F, Tornabene I, Tomarchio C, Drago M, Pavone G, Gorgone C, Barone C, Bianca S, Manzella L. In Silico Prediction of BRCA1 and BRCA2 Variants with Conflicting Clinical Interpretation in a Cohort of Breast Cancer Patients.Genes (Basel). 2024;15:943.
[RCA] [PubMed] [DOI] [Full Text][Cited by in RCA: 6][Reference Citation Analysis (0)]
Fraccascia B, Sodero G, Pane LC, Malavolta E, Gola C, Pane L, Paradiso VF, Nanni L, Rigante D, Cipolla C. Complete Androgen Insensitivity Syndrome in a Young Girl with Primary Amenorrhea and Suspected Delayed Puberty: A Case-Based Review of Clinical Management, Surgical Follow-Up, and Oncological Risk.Diseases. 2024;12:235.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in RCA: 7][Reference Citation Analysis (1)]
Hage M, Drui D, Francou B, Mercier S, Guiochon-Mantel A, Belaisch-Allart J, Péréon Y, Cazabat L, De Mazancourt P, Raffin-Sanson ML. Structural analysis of the impact of a novel androgen receptor gene mutation in two adult patients with mild androgen insensitivity syndrome.Andrologia. 2021;53:e13865.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 2][Cited by in RCA: 5][Article Influence: 0.8][Reference Citation Analysis (0)]
Tirrito L, Casciani F, Lomuscio S, Nardone AM, Di Rosa C, Libotte F, Ruta R, Novelli A, Rizzo G, Marchionni E, Novelli G. Familial androgen insensitivity syndrome caused by the AR N706S variant: case report and systematic literature review.J Sex Gender Specif Med. 2024;10:155-163.
[PubMed] [DOI] [Full Text]
Patjamontri S, Lucas-Herald AK, Bryce J, van den Akker E, Cools M, Globa E, Guerra-Junior G, Hiort O, Hofman P, Holterhus PM, Hughes IA, Juul A, Nordenstrom A, Russo G, Stancampiano MR, Seneviratne SN, Tadokoro-Cuccaro R, Thankamony A, Weintrob N, Zelinska N, Ahmed SF. Gynecomastia and Its Management In Boys With Partial Androgen Insensitivity Syndrome.J Clin Endocrinol Metab. 2025;110:e2018-e2025.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 3][Cited by in RCA: 6][Article Influence: 6.0][Reference Citation Analysis (0)]
Navarro G, Xu W, Jacobson DA, Wicksteed B, Allard C, Zhang G, De Gendt K, Kim SH, Wu H, Zhang H, Verhoeven G, Katzenellenbogen JA, Mauvais-Jarvis F. Extranuclear Actions of the Androgen Receptor Enhance Glucose-Stimulated Insulin Secretion in the Male.Cell Metab. 2016;23:837-851.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 114][Cited by in RCA: 140][Article Influence: 14.0][Reference Citation Analysis (0)]
Andrisse S, Feng M, Wang Z, Awe O, Yu L, Zhang H, Bi S, Wang H, Li L, Joseph S, Heller N, Mauvais-Jarvis F, Wong GW, Segars J, Wolfe A, Divall S, Ahima R, Wu S. Androgen-induced insulin resistance is ameliorated by deletion of hepatic androgen receptor in females.FASEB J. 2021;35:e21921.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 5][Cited by in RCA: 35][Article Influence: 7.0][Reference Citation Analysis (0)]
Lai Y, Ramírez-Pardo I, Isern J, An J, Perdiguero E, Serrano AL, Li J, García-Domínguez E, Segalés J, Guo P, Lukesova V, Andrés E, Zuo J, Yuan Y, Liu C, Viña J, Doménech-Fernández J, Gómez-Cabrera MC, Song Y, Liu L, Xu X, Muñoz-Cánoves P, Esteban MA. Multimodal cell atlas of the ageing human skeletal muscle.Nature. 2024;629:154-164.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 138][Cited by in RCA: 136][Article Influence: 68.0][Reference Citation Analysis (1)]
Audi L, Ahmed SF, Krone N, Cools M, McElreavey K, Holterhus PM, Greenfield A, Bashamboo A, Hiort O, Wudy SA, McGowan R; The EU COST Action. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 ‘DSDnet’.Eur J Endocrinol. 2018;179:R197-R206.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 129][Cited by in RCA: 113][Article Influence: 14.1][Reference Citation Analysis (0)]
Nordenström A, Ahmed SF, van den Akker E, Blair J, Bonomi M, Brachet C, Broersen LHA, Claahsen-van der Grinten HL, Dessens AB, Gawlik A, Gravholt CH, Juul A, Krausz C, Raivio T, Smyth A, Touraine P, Vitali D, Dekkers OM. Pubertal induction and transition to adult sex hormone replacement in patients with congenital pituitary or gonadal reproductive hormone deficiency: an Endo-ERN clinical practice guideline.Eur J Endocrinol. 2022;186:G9-G49.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 9][Cited by in RCA: 73][Article Influence: 18.3][Reference Citation Analysis (1)]
European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD).J Hepatol. 2024;81:492-542.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 1461][Cited by in RCA: 1394][Article Influence: 697.0][Reference Citation Analysis (6)]
Gallardo-Durán KG, De la Fuente-Cortez BE, Villarreal-Benavides TC, García-Vielma C. [Complete androgen insensitivity syndrome: diagnosis and multidisciplinary management].Rev Med Inst Mex Seguro Soc. 2023;61:117-122.
[PubMed] [DOI]
Salles J, Chanet A, Guillet C, Vaes AM, Brouwer-Brolsma EM, Rocher C, Giraudet C, Patrac V, Meugnier E, Montaurier C, Denis P, Le Bacquer O, Blot A, Jourdan M, Luiking Y, Furber M, Van Dijk M, Tardif N, Yves Boirie Y, Walrand S. Vitamin D status modulates mitochondrial oxidative capacities in skeletal muscle: role in sarcopenia.Commun Biol. 2022;5:1288.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in RCA: 50][Reference Citation Analysis (0)]
Birnbaum W, Marshall L, Werner R, Kulle A, Holterhus PM, Rall K, Köhler B, Richter-Unruh A, Hartmann MF, Wudy SA, Auer MK, Lux A, Kropf S, Hiort O. Oestrogen versus androgen in hormone-replacement therapy for complete androgen insensitivity syndrome: a multicentre, randomised, double-dummy, double-blind crossover trial.Lancet Diabetes Endocrinol. 2018;6:771-780.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 21][Cited by in RCA: 35][Article Influence: 4.4][Reference Citation Analysis (1)]
Kohrt SE, Novak EJ, Tapadar S, Wu B, Strope J, Asante Y, Kim H, Chang MS, Gurdak D, Khalil A, Rood M, Raftery E, Stavreva D, Nguyen HM, Brown LG, Ramser M, Peer C, Meyers WM, Aboreden N, Chakravortee M, Sallari R, Nelson PS, Kelly KK, Graham TGW, Darzacq X, Figg WD, Oyelere AK, Corey E, Adelaiye-Ogala R, Gryder BE. Small-molecule disruption of androgen receptor-dependent chromatin clusters.Proc Natl Acad Sci U S A. 2024;121:e2406239121.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 4][Cited by in RCA: 4][Article Influence: 2.0][Reference Citation Analysis (0)]
Austregésilo de Athayde De Hollanda Morais B, Martins Prizão V, de Moura de Souza M, Ximenes Mendes B, Rodrigues Defante ML, Cosendey Martins O, Rodrigues AM. The efficacy and safety of GLP-1 agonists in PCOS women living with obesity in promoting weight loss and hormonal regulation: A meta-analysis of randomized controlled trials.J Diabetes Complications. 2024;38:108834.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 51][Cited by in RCA: 39][Article Influence: 19.5][Reference Citation Analysis (0)]
Kumari K, Kumar R, Memon A, Kumari B, Tehrim M, Kumari P, Shehryar M, Islam H, Islam R, Khatri M, Kumar S, Kumar A. Treatment with Testosterone Therapy in Type 2 Diabetic Hypogonadal Adult Males: A Systematic Review and Meta-Analysis.Clin Pract. 2023;13:454-469.
[RCA] [PubMed] [DOI] [Full Text][Cited by in RCA: 24][Reference Citation Analysis (0)]
Wechsung K, Marshall L, Jürgensen M, Wiegmann S, Kalender U, Brösamle M, Herrmann G, Hiort O, Janssen-Schmidchen G, Richter-Unruh A, Wabitsch M, Wunn C, Keil T, Neumann U, Stöckigt B. Structured care after a DSD diagnosis in childhood: a mixed methods evaluation of the Empower-DSD program.Front Pediatr. 2025;13:1488411.
[RCA] [PubMed] [DOI] [Full Text][Cited by in RCA: 5][Reference Citation Analysis (1)]
Konieczna J, Ruiz-Canela M, Galmes-Panades AM, Abete I, Babio N, Fiol M, Martín-Sánchez V, Estruch R, Vidal J, Buil-Cosiales P, García-Gavilán JF, Moñino M, Marcos-Delgado A, Casas R, Olbeyra R, Fitó M, Hu FB, Martínez-Gonzalez MÁ, Martínez JA, Romaguera D, Salas-Salvadó J. An Energy-Reduced Mediterranean Diet, Physical Activity, and Body Composition: An Interim Subgroup Analysis of the PREDIMED-Plus Randomized Clinical Trial.JAMA Netw Open. 2023;6:e2337994.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in Crossref: 25][Cited by in RCA: 59][Article Influence: 19.7][Reference Citation Analysis (0)]
ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, Collins BS, Hilliard ME, Isaacs D, Johnson EL, Kahan S, Khunti K, Leon J, Lyons SK, Perry ML, Prahalad P, Pratley RE, Seley JJ, Stanton RC, Gabbay RA; on behalf of the American Diabetes Association. 8. Obesity and Weight Management for the Prevention and Treatment of Type 2 Diabetes: Standards of Care in Diabetes-2023.Diabetes Care. 2023;46:S128-S139.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 2][Cited by in RCA: 157][Article Influence: 52.3][Reference Citation Analysis (0)]
Mechanick JI, Apovian C, Brethauer S, Garvey WT, Joffe AM, Kim J, Kushner RF, Lindquist R, Pessah-Pollack R, Seger J, Urman RD, Adams S, Cleek JB, Correa R, Figaro MK, Flanders K, Grams J, Hurley DL, Kothari S, Seger MV, Still CD. Clinical Practice Guidelines For The Perioperative Nutrition, Metabolic, And Nonsurgical Support Of Patients Undergoing Bariatric Procedures - 2019 Update: Cosponsored By American Association Of Clinical Endocrinologists/American College Of Endocrinology, The Obesity Society, American Society For Metabolic & Bariatric Surgery, Obesity Medicine Association, And American Society Of Anesthesiologists - Executive Summary.Endocr Pract. 2019;25:1346-1359.
[RCA] [PubMed] [DOI] [Full Text][Cited by in Crossref: 272][Cited by in RCA: 281][Article Influence: 40.1][Reference Citation Analysis (0)]
Goodman M, Yacoub R, Getahun D, McCracken CE, Vupputuri S, Lash TL, Roblin D, Contreras R, Cromwell L, Gardner MD, Hoffman T, Hu H, Im TM, Prakash Asrani R, Robinson B, Xie F, Nash R, Zhang Q, Bhai SA, Venkatakrishnan K, Stoller B, Liu Y, Gullickson C, Ahmed M, Rink D, Voss A, Jung HL, Kim J, Lee PA, Sandberg DE. Cohort profile: pathways to care among people with disorders of sex development (DSD).BMJ Open. 2022;12:e063409.
[RCA] [PubMed] [DOI] [Full Text] [Full Text (PDF)][Cited by in RCA: 7][Reference Citation Analysis (2)]
Footnotes
Provenance and peer review: Invited article; Externally peer reviewed.
Peer-review model: Single blind
Specialty type: Endocrinology and metabolism
Country of origin: China
Peer-review report’s classification
Scientific Quality: Grade B, Grade C
Novelty: Grade A, Grade C
Creativity or Innovation: Grade A, Grade C
Scientific Significance: Grade B, Grade B
Open Access: This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
P-Reviewer: Tung TH, PhD, Associate Professor, Director, Taiwan; Vaitsopoulou CI, MD, PhD, Greece; Wang L, PhD, Director, Research Dean, China S-Editor: Bai Y L-Editor: A P-Editor: Lei YY